
通过对670例Dravet综合征(DS)患儿进行随访,对其预后进行总结。
收集2005年2月至2016年8月在北京大学第一医院儿科就诊的DS患儿,建立临床资料登记表,完善基因检查。通过门诊复诊及电话随访的方式对DS患儿的预后进行随访。
670例DS患儿中,存在SCN1A突变者556例(556/670例,83.0%),随访608例(90.7%,608/670例),失访62例(失访率为9.3%,62/670例);末次随访中位年龄8岁5个月。82例(82/608例,13.5%)发作曾控制1年以上,中位随访年龄9岁2个月。其中38例再次出现发作(38/82例,46.3%),主要诱发因素为发热(34例)或漏服抗癫痫药物(2例)。分析发作曾控制1年以上的相关因素,发现携带SCN1A错义突变者、遗传性突变者、年龄相对较大者发作控制相对较好。随访的608例患儿中,死亡25例(25/608例,4.1%),发生死亡的中位年龄为4岁;12例因病程中出现长时间的癫痫持续状态,并多器官衰竭死亡。7例为可能的癫痫猝死,2例为呕吐窒息死亡,1例外伤后死亡,余3例死因不详。
DS为难治性癫痫,但少数患儿发作可控制1年以上,携带SCN1A错义突变者、遗传性突变者、年龄相对较大者发作控制相对较好。DS病死率高,死亡原因主要为癫痫持续状态后多脏器衰竭和可能的癫痫猝死。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Dravet综合征(DS)为婴儿期起病的难治性癫痫综合征,多数患儿预后不良,发作难以控制,到成年期常仍有发作,多数患儿智力发育中重度落后,且病死率高,文献报道病死率为10.1%~15.0%[1,2]。随年龄增长,多数DS患儿发作的热敏感性降低,发作次数有减少趋势。本研究对670例DS患儿进行长期随访,对发作控制1年以上患儿的临床特点及相关因素进行分析,并对死亡患儿的原因进行分析,以提高儿科临床医师及患儿家长对本病预后的认识,指导其对患儿进行长期管理。
收集2005年2月至2016年8月在北京大学第一医院儿科就诊的670例DS患儿,其中男377例,女293例。至末次随访时,患儿年龄为3岁7个月~27岁(中位年龄8岁5个月)。通过门诊复诊及电话随访,记录患儿发作、治疗及智力运动发育情况等。根据发作控制情况分为相对良好组(发作曾控制1年以上)和相对欠佳组(发作未控制1年以上)。本研究通过北京大学第一医院医学伦理委员会批准[批准文号:2012(453)],患儿监护人均签署知情同意书。
采用SPSS 23.0软件进行统计分析。计数资料采用例数和百分数(%)描述;正态分布的计量资料采用 ±s表示;偏态分布的计量资料采用中位数(范围)表示。分析其相关影响因素,2组起病年龄及末次随访年龄比较采用t检验。2组性别、SCN1A突变类型、突变来源、既往发生癫痫持续状态(SE)次数比较采用χ2检验。P<0.05为差异有统计学意义。
±s表示;偏态分布的计量资料采用中位数(范围)表示。分析其相关影响因素,2组起病年龄及末次随访年龄比较采用t检验。2组性别、SCN1A突变类型、突变来源、既往发生癫痫持续状态(SE)次数比较采用χ2检验。P<0.05为差异有统计学意义。
670例DS患儿中,失访62例(9.3%)。670例DS患儿中,SCN1A突变阳性者556例(83.0%)。分析501例患儿父母SCN1A基因突变,证实患儿为新生突变451例(90.0%),遗传性突变50例(10.0%)。PCDH19突变者6例,GABRA1杂合突变3例,GABRG2杂合突变2例,GABRB2杂合突变2例,TBC1D24复合杂合突变2例,ALDH7A1复合杂合突变2例,SCN2A杂合突变1例。
670例DS患儿中,随访608例(90.7%)。其中男336例,女272例,随访中位年龄8岁5个月。其中161例(161/608例,26.5%)患儿发作随年龄增长逐渐减少,发作减少中位年龄为5岁(4~15岁)。79例通过调整抗癫痫药物(AEDs),仅在发热情况下出现发作。82例发作曾控制1年以上(82/608例,13.5%),其中发作控制2年以上者34例,3年以上者15例,4年以上者4例,5年以上者2例(该2例患儿均已减停AEDs)。82例发作曾控制1年以上的患儿中,38例之后再次出现发作(38/82例,46.3%),再次出现发作的年龄为3岁3个月~15岁(中位年龄7岁)。诱发因素为发热者34例,漏服药物者2例,无明确发作诱因者2例。其中24例患儿目前1年发作1~3次(多因发热诱发),7例1年发作4~10次,7例1个月发作1~2次。
发作曾控制1年以上的82例患儿中,男46例,女36例;SCN1A突变阳性者57例(69.1%),阴性者25例(30.9%)。SCN1A突变阳性57例中,错义突变35例(61.4%),截断突变15例(26.3%,包括无义突变9例,移码突变6例),剪切位点突变4例(7.0%),片段缺失3例(5.3%);遗传性突变13例(22.8%),新生突变44例(77.2%)。SCN1A突变阴性的25例患儿中,GABRB2突变者2例,GABRA1突变者2例,ALDH7A1复合杂合突变者2例,GABRG2突变者1例,PCDH19突变者1例。末次随访年龄2岁9个月~17岁(中位随访年龄9岁2个月);发作开始减少的年龄为4~10岁(平均发作减少年龄5.6岁);起病年龄3~11个月(平均起病年龄6个月),病程中曾出现肌阵挛发作者43例,出现不典型失神者22例。出现SE≥3次者57例(69.5%)。2药联用44例,3药联用35例,4药联用3例。其中以丙戊酸钠联合左乙拉西坦(21例)最为常见,丙戊酸钠、左乙拉西坦联合托吡酯者15例、丙戊酸钠联合托吡酯14例。34例加用托吡酯有效,15例加用丙戊酸钠治疗有效,12例加用氯硝西泮治疗后发作减少,10例生酮饮食治疗有效,9例加用左乙拉西坦后发作减少,2例加用唑尼沙胺后发作控制(图1)。
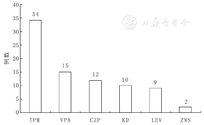
注:TPM:托吡酯;VPA:丙戊酸;CZP:氯硝西泮;KD:生酮饮食;LEV:左乙拉西坦;ZNS:唑尼沙胺 TPM:Topiramate;VPA:Valproate acid;CZP:Clonazepam;KD:ketogenic diet;LEV:Levetiracetam;ZNS:Zonisamide
进一步分析DS患儿发作控制相对良好的相关因素(表1),发现携带SCN1A错义突变者、遗传性突变者、年龄相对较大者发作控制相对较好(均P<0.05)。其发作控制情况与起病年龄,既往SE次数无明显相关性(均P>0.05)。
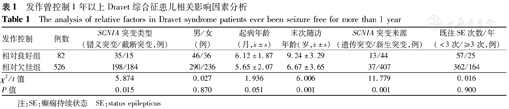
发作曾控制1年以上Dravet综合征患儿相关影响因素分析
The analysis of relative factors in Dravet syndrome patients ever been seizure free for more than 1 year
发作曾控制1年以上Dravet综合征患儿相关影响因素分析
The analysis of relative factors in Dravet syndrome patients ever been seizure free for more than 1 year
| 发作控制 | 例数 | SCN1A突变类型(错义突变/截断突变,例) | 男/女(例) | 起病年龄(月, ±s) ±s) | 末次随访年龄(岁, ±s) ±s) | SCN1A突变来源(遗传突变/新生突变,例) | 既往SE次数/年(<3次/≥3次,例) |
|---|---|---|---|---|---|---|---|
| 相对良好组 | 82 | 35/15 | 46/36 | 6.12±1.87 | 9.24±3.29 | 13/44 | 57/25 |
| 相对欠佳组 | 526 | 198/184 | 290/236 | 5.65±2.07 | 6.67±3.65 | 37/407 | 362/164 |
| χ2/t值 | 5.874 | 0.027 | 1.936 | 6.006 | 11.779 | 0.016 | |
| P值 | 0.015 | 0.870 | 0.051 | 0.001 | 0.001 | 0.900 |
注:SE:癫痫持续状态 SE:status epilepticus
随访608例DS患儿中,死亡25例(4.1%)。发生死亡的年龄为15个月~12岁,中位年龄为4岁。其中12例因病程中出现SE,发作时间长,出现急性脑病,并多器官衰竭死亡;7例为可能的癫痫猝死(SUDEP),其中2例在北京大学第一医院就诊,有详细住院资料。此2例患儿均在全面强直阵挛发作(GTCS)1~2 min后,突然出现呼吸心跳骤停,经及时抢救后恢复自主心率,自主呼吸未恢复,床旁脑电图提示持续低电压,家属放弃治疗后死亡;另有2例均于睡眠中发生,被监护人发现时已无生命体征,可见呕吐物,不除外呕吐后窒息所致,发生呕吐前是否出现抽搐发作情况不详。1例意外坠楼死亡。另3例具体死因不详。25例死亡患儿中,11例携带SCN1A错义突变,10例携带SCN1A截断突变(6例为无义突变,4例为移码突变),1例携带SCN1A剪切位点突变,1例携带SCN1A大片段缺失,2例未携带SCN1A基因变异。7例可能的SUDEP患儿中,2例携带SCN1A错义突变,2例携带SCN1A无义突变,1例携带SCN1A大片段缺失,2例未携带SCN1A基因变异。
随访中家长诉患儿有多动表现321例(52.8%)。22例存在孤独症样表现,中位年龄9岁6个月(6~19岁),7例无自主性语言表达,4例有刻板动作和行为,其他表现为对话时无眼神交流,"沉浸在自己的世界中"。67例(11.0%)存在睡眠障碍,表现为入睡困难、浅睡时间长、易惊醒。3例患儿出现帕金森样症状,表现为持物手抖、震颤,紧张时加重,末次随访年龄分别为10岁、14岁和17岁。
随访的DS患儿中,部分运动功能受累,其中355例(58.4%)存在共济失调。4例在发生SE后导致惊厥性脑损伤,恢复期遗留偏瘫。8例存在蹲伏步态,表现为膝关节屈曲内收,无法伸直,影响行走,出现蹲伏步态的中位年龄为10.5岁(6~23岁),均携带SCN1A突变,其中错义突变5例,无义突变2例,剪切位点突变1例。
DS为婴儿期起病的难治性癫痫综合征。其主要致病基因为SCN1A,该基因突变检出率约为80%,本研究670例DS患儿中,SCN1A突变阳性者556例(556/670例,83.0%)。DS患儿其他少见致病基因包括PCDH19、GABRG2、SCN2A、GABRA1、GABRB2、TBC1D24和ALDH7A1[3]。
DS为难治性癫痫综合征,多数患儿发作控制欠佳。目前中国大陆可用的AEDs有丙戊酸、托吡酯、左乙拉西坦和氯硝西泮。文献报道其他可用于DS的AEDs有氯巴占、司替戊醇、唑尼沙胺等[4,5,6,7]。新药芬氟拉明及大麻二酚对于DS的疗效观察目前尚处于临床试验阶段[6,7]。DS患儿多采用多种AEDs联合应用,但多数发作控制欠佳。钠离子通道阻滞剂卡马西平、奥卡西平和拉莫三嗪等药物可加重发作。目前中国大陆尚无氯巴占、司替戊醇、芬氟拉明和大麻二酚等药物。
虽然DS为难治性癫痫综合征,多数患儿发作控制欠佳,但也有少数患儿随年龄增长发作逐渐减少,极少数患儿发作可完全控制[8]。文献报道少数DS患儿发作可控制1年以上,分析其相关因素,发现既往SE次数≤3次、脑电图异常放电逐渐消失常提示发作相对易控制,还可能与添加托吡酯和/或司替戊醇治疗有关;但与SCN1A突变类型、性别无关[8,9,10]。因此早期诊断并添加有效AEDs,对控制发作有帮助。本研究随访到608例DS患儿,82例发作曾控制1年以上,其中2例发作控制5年后已停用AEDs。添加有效的药物主要有托吡酯、丙戊酸和氯硝西泮。分析其相关因素,发现SCN1A为错义突变者、遗传性突变者和年龄较大者发作相对容易控制。发作曾控制1年以上的SCN1A突变阴性的患儿其致病基因可为GABRG2、GABRB2、GABRA1、PCDH19及ALDH7A1。在82例患儿中,少数可在发热或漏服药物后再次出现发作,但发作较稀少。年龄越小,出现再次发作的可能性越大。本研究发现,82例患儿发作控制相对较好,提示并非所有的DS患儿发作都难以控制。因此,在临床工作中,应尽早给予有效治疗,并告知患儿及家属,帮助其树立对抗疾病的信心,提高用药依从性。
DS被国际抗癫痫联盟(ILAE)归为癫痫性脑病之一,绝大多数患儿1岁前发育正常,1岁后逐渐出现发育停滞甚至倒退[8]。DS患儿随年龄增长可出现共济失调,锥体束征,少数可有蹲伏步态[11]。本研究随访到608例DS患儿,随访中位年龄为8岁5个月,58.4%的患儿存在共济失调,8例出现蹲伏步态,影响行走。研究认为,SCN1A突变导致钠离子通道功能减低,可能影响轴索功能导致步态异常[12,13]。本组8例有蹲伏步态的患儿均携带SCN1A突变,突变类型包括错义突变、无义突变和剪切位点突变。蹲伏步态与其SCN1A突变类型无关。本课题组计划对该8例患儿随访头颅MRI及功能影像学检查,以对DS患儿蹲伏步态的发生机制进一步研究。
癫痫患儿可出现其他神经及精神系统疾病,称之为癫痫共患病,如注意力缺陷多动障碍、孤独症谱系障碍(ASD)等[14,15,16]。ASD是一组以社交障碍、语言交流障碍、兴趣或活动范围狭窄及重复刻板行为为主要特征的神经发育性障碍[17]。本研究在门诊复诊的DS患儿中也发现,少数患儿发作控制尚可,但缺少眼对眼交流,很少或无自主性语言表达(非药物不良反应或癫痫性脑病导致)。对其发放社会反应量表进行初步评估,发现可能存在孤独症倾向,给予针对性训练,少数患儿症状可有改善。
Sakauchi等[1]随访623例DS患者,病死率为10.1%(63/623例),3~7岁为死亡的高峰年龄,死因包括SUDEP(31/63例,49.2%),SE后多脏器衰竭(21/63例,33.3%),或发作时溺水等意外死亡等。治疗用药和发作次数与死亡无明显相关性[1]。SCN1A基因突变可导致自主神经功能障碍,以及心肌细胞钠离子内流增加,QT间期延长,心率变异性(HRV)减低,从而导致SUDEP[18]。SUDEP是指癫痫患者突然发生的、缺乏合理的解剖学及毒理学证据的死亡。研究表明,SUDEP发生的高危因素有起病年龄小、GTCS发作频繁、病程长等因素[19,20]。DS患儿起病年龄在1岁以内,发作控制欠佳,80%的患儿携带SCN1A基因突变,是发生SUDEP的高危人群。本研究随访到608例患儿,25例(4.1%)死亡,死亡中位年龄为4岁。其中12例死于SE,7例死于可能的SUDEP,2例可能为呕吐后窒息导致,1例因看护疏漏意外坠楼身亡,余3例死因不详。7例可能的SUDEP患儿中,5例携带SCN1A突变,包括错义突变、无义突变及大片段缺失,因例数较少,目前未发现DS患儿SCN1A突变类型与其是否会发生SUDEP存在相关性。本研究病死率低于国际报道,考虑与随访时间短有关,少数失访患儿不除外死亡的可能。DS的病死率高于癫痫一般人群,因此DS患儿应注意避免高热,加强看护,发作时及时止惊,减少SE的发生。国外报道建议对DS患儿常规进行心功能评估,可通过佩戴心率监测仪,如有发作或呼吸停止,予以警报提醒,以便及时发现异常,给予干预[21]。
综上,DS为难治性癫痫综合征,少数患儿经积极治疗后发作可控制1年以上;SCN1A突变为错义突变者、遗传性突变者,年龄较大者,发作相对易控制;DS患儿病死率高于普通癫痫患儿,注意发生SE时尽早止惊,避免感染,控制GTCS发作,以及加强监护等,可减少其死亡的发生风险。
所有作者均声明不存在利益冲突

























