
本文报告了1例染色体核型为46,XX,Y染色体性别决定基因(sex-determining region on the Y chromosome,SRY基因)阳性的孕妇及其女性胎儿。孕12周+6时,超声显示胎儿颈项透明层增厚,羊水染色体核型和定量荧光聚合酶链反应检测结果显示胎儿核型为46,XX,SRY基因阳性。但B超显示胎儿社会性别为女性,与46,XX,SRY阳性男性综合征表型不一致。通过荧光原位杂交技术分析结果发现1条X染色体长臂2区8带(Xq28)包含Y染色体短臂1区1带3亚带(Yp11.3)片段。同时,染色体微阵列分析技术检出Yp11.31p11.2的片段重复,长度约1 Mb,证实了胎儿X染色体含有部分Y染色体片段,与孕妇本人一致。经遗传咨询,胎儿父母选择继续妊娠至足月分娩,随访未见异常。


孕妇32岁,社会性别为女性,因孕12周+6宫内超声测量胎儿颈项透明层增厚(7.5 mm),于2019年4月10日(孕14周)来珠海市妇幼保健院就诊,要求对胎儿染色体异常风险进行评估。该孕妇为孕2产1,2014年生育一男孩,表型及智力均正常,体健。孕妇否认家族遗传病史,否认近亲婚配。配偶体格检查未见明显异常。孕妇体格检查:身高160.5 cm,体重56 kg,智力发育正常,无喉结,双侧乳房发育饱满,外阴发育正常,超声可探测到子宫及双侧卵巢附件。2019年4月19日(孕15周+2)检测性激素水平未见异常。
取得孕妇知情同意后,于孕22周行羊膜腔穿刺产前诊断。经染色体核型分析,胎儿的染色体核型为46,XX,Y染色体性别决定基因(sex-determining region on the Y chromosome, SRY)基因阳性。取得孕妇及家属知情同意后,又分别抽取孕妇、配偶及其子静脉血3 ml提取DNA,以及肝素抗凝血5 ml送本院检验科(医学遗传研究所)行染色体核型分析。经检测,孕妇和胎儿的核型一致,均为46,XX,SRY基因阳性。其子和配偶核型为46,XY,与社会性别一致,未见异常。而非整倍体快速检测的荧光定量聚合酶链反应(quantitative fluorescent-polymerase chain reaction, QF-PCR)检测结果显示胎儿与孕妇一致。见图1。
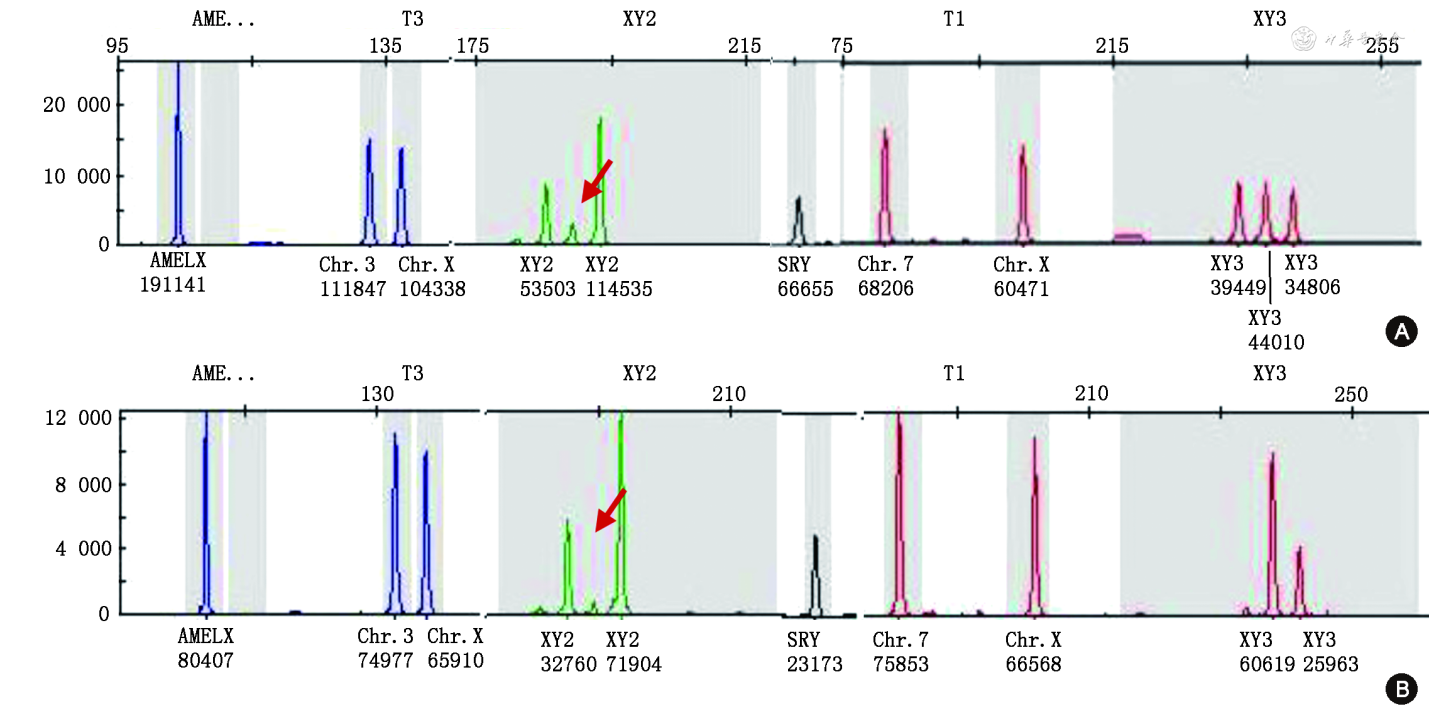
注:AMELX为融合蛋白基因,T1为7号和X染色体基因座,T3为3号和X染色体基因座
随后行染色体荧光原位杂交(fluorescence in situ hybridization, FISH)分析,发现孕妇和胎儿有部分Y染色体短臂片段易位至X染色体长臂,1条X染色体长臂2区8带(Xq28)包含Y染色体短臂1区1带3亚带(Yp11.3)片段,即Yp11.3-Xq28(图2)。进一步行染色体微阵列分析(chromosomal microarray analysis, CMA),检出孕妇和胎儿均存在一段单拷贝Yp11.31p11.2,长度约1 Mb,包含RPS4Y1、SRY、ZFY和TGIF2LY等4个在线人类孟德尔遗传基因(图3),验证了QF-PCR的结果。同时,Y染色体微缺失基因检测和SRY基因DNA测序结果表明,孕妇及胎儿Y染色体均缺失AZFa、AZFb及AZFc区,但SRY基因阳性(图4)。SRY基因DNA测序结果显示孕妇、胎儿、配偶及其子SRY基因未出现突变。
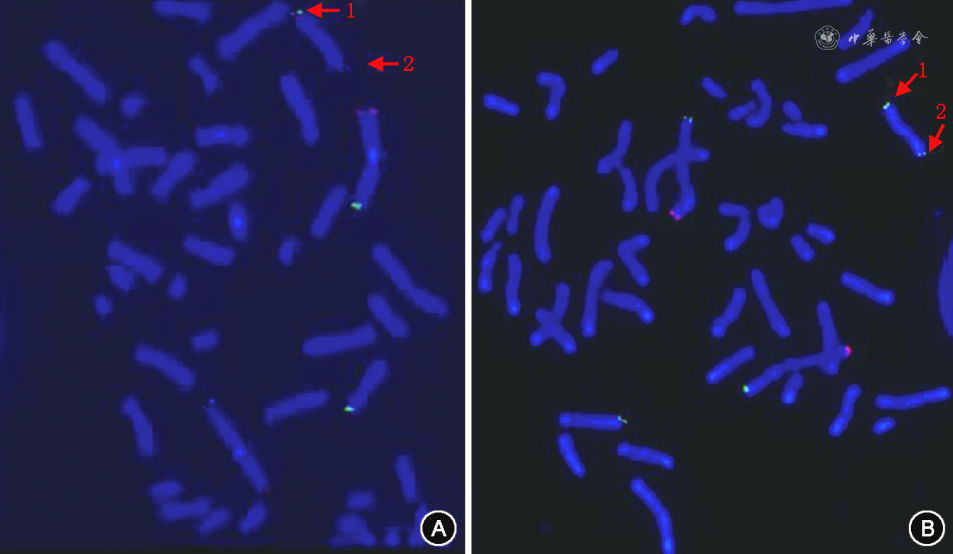

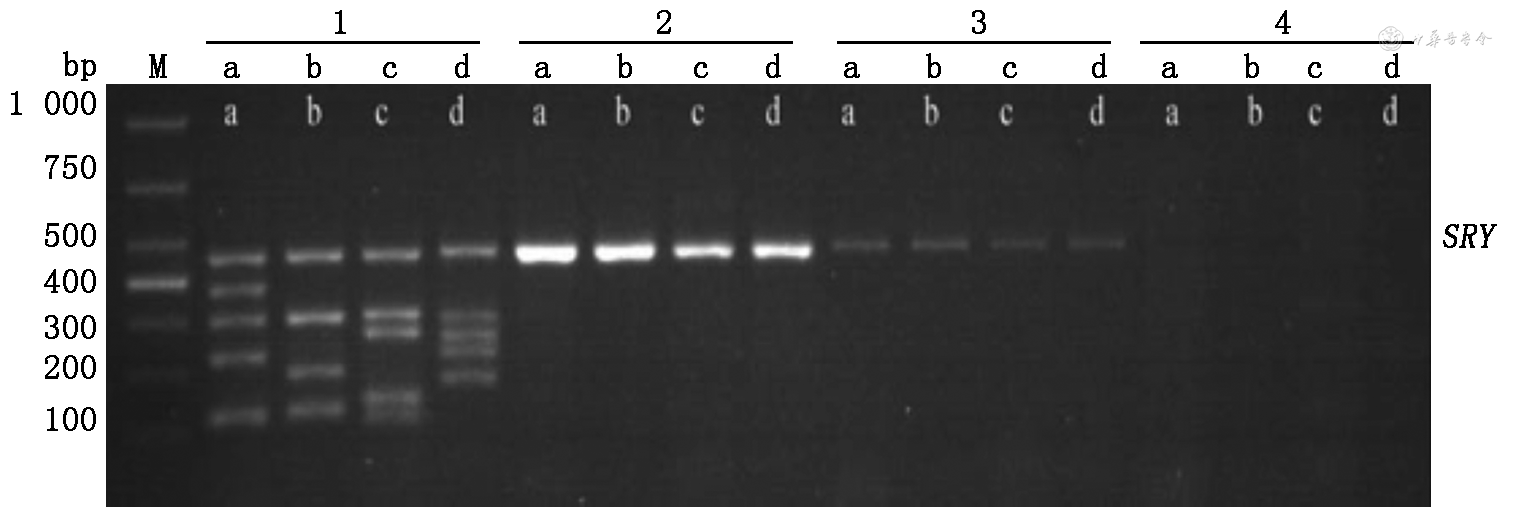
注:M:参照标准;1:正常男性对照;2:本例孕妇;3:本例胎儿;4.正常女性对照;SRY:Y染色体性别决定基因(sex-determining region on the Y chromosome);a、b、c、d分别代表AZFa、AZFb、AZFc和AZFd区
经遗传咨询,考虑到胎儿包含Yp的X染色体来源于母亲,且性别表型与母亲一致,重复的片段无致病性,因此胎儿父母选择继续妊娠。该胎儿于孕39周+1经阴道分娩出生,生后体格检查未见明显异常。2020年5月电话随访时,其生长发育和体格检查未见异常。
46,XX,SRY基因阳性个体社会性别多表现为男性,称为男性综合征[1, 2]。而本例孕妇社会性别与胎儿核型均为女性,与检索到的临床报道[3, 4, 5]不一致。性发育包括性别决定和性别分化等阶段,是在一系列的基因作用下,性腺向睾丸或者卵巢分化的过程。早期研究发现,人类Y染色体上存在睾丸决定因子(testis determining factor, TDF)。现已研究表明SRY基因是TDF最佳候选基因。SRY基因缺失、突变、易位均可导致性别发育异常[6]。46,XX,SRY基因阳性个体常常表现为男性,故称为男性综合征,临床表现为第二性征不足,常见阴茎小、睾丸小、身材矮小,常常因为婚后不育就诊而被发现[7, 8]。该病常见原因为Yp-Xp末端易位,也有极少SRY与常染色体相互易位,导致XX男性的报道[9, 10],而本例因检出Yp-Xq而具有特殊性。
本病例QF-PCR、染色体核型分析显示孕妇和胎儿染色体核型为46,XX,但SRY基因阳性,与常见的临床报道[11]不符。有报道46,XX,SRY(+)女性存在两性畸形、阴蒂肥大等发育异常[12]。而本报道中孕妇性别表型未见异常,且已生育一子。46,XX,SRY(+)常见临床表型为男性,多由Yp-Xp末端易位所致。本例与常见临床报道不一致的原因存在以下几种可能。一是FISH分析结果显示孕妇和胎儿均存在衍生X染色体,含SRY基因的Y染色体片段,即Yp-Xq。而基因的表达受时间和空间因素的影响,推测SRY基因移位至X染色体长臂,空间位置发生了改变,导致SRY基因不能表达,从而表现女性性别。二是可能存在X染色体失活机制,导致SRY基因表达受到抑制。X染色体失活过程受X染色体失活中心(X chromosome inactivation center,XIC)编码的多个长非编码RNA调控,包括Xist、Tsix等结合到XIC上,进而富集更多的染色体修饰相关复合物,促使异染色质构象的形成。Lyon假说认为X染色体失活是随机的,但也有研究表明X染色体可能为非随机失活,可能优先失活有突变的X染色体[13]。因此本案例中是否优先失活存在Y染色体片段的衍生X染色体而导致SRY基因不表达,有待于进一步研究。三是可能存在与性别决定相关的其他基因发生突变。CMA结果显示孕妇和胎儿均存在单拷贝Yp11.31p11.2的片段重复,包含了SRY基因。QF-PCR和FISH结果也显示存在SRY基因。虽然本单位对SRY基因进行了测序,未发现突变,但可能存在其他与性别决定相关的基因如SOX3、SOX9等发生突变,使得该胎儿虽然存在SRY基因却不能表现为男性性别。细胞和分子遗传学分析结果显示胎儿衍生的Yp-Xq染色体来源于母亲,不是新发突变,但该易位方式比较罕见。由于没有对更多的家庭成员进行分析,该易位型性染色体是否由母系成员遗传,有待进一步研究。
综上所述,性别发育异常不是单一的一种疾病,这类疾病有着共同的临床特点,性发育不全或两性畸形,也有不同的细胞及分子遗传背景。本例孕妇染色体核型为46,XX,SRY基因阳性,但社会性别为女性,与临床常见报道并不一致。由于46,XX,SRY阳性男性综合征表型的临床特征具有一定的隐蔽性,因此,对于此类患者应结合其社会性别、染色体核型、FISH分析、SRY基因的检测,特别是新兴的染色体微阵列分析技术,为患者的诊断以及遗传咨询提供科学的依据,并为生育提供指导意见。

























