版权所有,未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别申明,本刊刊出的所有文章不代表中华医学会和本刊编辑委员会的观点。
光盘如有质量问题,请向编辑部调换
男性乳腺发育症是指男性乳腺基质及腺管异常增多、乳腺外形增大的乳腺良性疾病,约占男性乳腺疾病的90%,主要原因为雌二醇与雄激素的比值增加[1,2]。XX男性综合征,是两性畸形中较少见的类型,其性腺表型为男性睾丸,而染色体性别却是女性。患者多因"外生殖器异常、不育、男性乳腺发育症"就诊,染色体检查为46,XX[3,4]。东莞市人民医院于2016年9月诊治1例以双侧乳腺发育为特点的XX男性综合征,笔者结合相关文献对该病的临床特点、诊断、鉴别诊断及治疗进行分析探讨。
患者社会性别男性,27岁,因"双侧乳腺发育11年余"于2016年9月10日入院。患者11年余前出现双侧乳腺发育,给予中药治疗(具体用药不详)1个月后自行消退,半年前再次出现双侧乳腺发育,伴性欲及性功能稍减退。无泌乳、乳腺疼痛等。既往体健,否认长期用药史。患者为足月顺产,无宫内窒息病史,现未婚未育。父亲168.0 cm,母亲150.0 cm,母亲怀孕2次,生产1次,流产1次(G2P1A1),流产原因不详,无兄弟姐妹。否认父母近亲结婚,否认家族遗传疾病史。查体:身高163.5 cm,体重68.5 kg,指尖距161.0 cm,未见喉结及胡须,腋毛少;双侧乳房外上象限可触及腺体,Tanner分期Ⅱ期[5],挤压乳头无溢液;阴毛呈PH4期,男性外生殖器,阴茎长度5.5 cm,周径5.0 cm,左侧睾丸3 ml,右侧睾丸5 ml,生殖器发育G2至G3期。未见外阴、阴蒂、阴道等女性生殖器。实验室检查:睾酮2.82 nmol/L(正常范围:6.07~27 nmol/L),促黄体生成素19.54 U/L (正常范围:1.24~8.62 U/L),卵泡刺激素33.51 U/L(正常范围:1.27~19.26 U/L)。催乳素、生长激素、甲状腺激素、皮质醇及促肾上腺皮质激素、脱氢表雄酮、17-羟孕酮、肝肾功能检查未见异常。精液检测:镜下可见大量上皮细胞,离心后未见精子。睾丸超声显示:双侧睾丸体积偏小,左侧14 mm×7 mm、右侧15 mm×8 mm(正常范围:长径35~45 mm,宽径20~30 mm)。
乳腺超声显示双侧乳头后方皮下类腺体组织,左侧42 mm×9 mm、右侧50 mm×8 mm,符合乳腺发育声像。盆腔超声未见子宫及附件。泌尿系统超声显示前列腺偏小,约为20 mm×18 mm×18 mm。
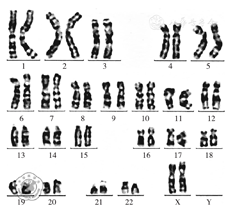
外周血染色体检测为46,XX(图1)。采用PCR及双向DNA测序方法检测SRY基因,根据SRY基因序列(NG_011751.1),应用Primer Premier 5软件设计特异性引物进行PCR扩增。PCR扩增产物经1.5%琼脂糖凝胶中电泳15 min后,溴化乙锭染色,于凝胶成像系统中判断结果。判断标准:电泳结果观察到1 146 bp的PCR产物,每次实验均设阴性(正常女性)、阳性(正常男性)作为质控对照。为进一步验证PCR产物结果的特异性,PCR扩增产物经DNA纯化试剂盒纯化后,送上海生物工程有限公司进行双向DNA测序(ABI 3730XL自动测序仪)。测序结果采用Chromas软件进行分析,并与标准参考序列进行Blast比对,证实该PCR产物为SRY基因(图2、图3)。

注:1为DNA标志物,2为阳性对照(正常男性),3为阴性对照(正常女性),4为患者;该患者SRY基因检测阳性

注:PCR产物双向测序结果与SRY基因参考序列相符
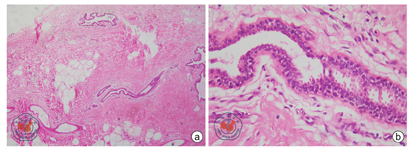
患者诊断XX男性综合征明确。给予十一酸睾酮补充雄激素治疗(每次40 mg,每晚1次),建议患者尽早行乳腺腺体切除术。2017年9月5日,患者在东莞市人民医院乳腺外科行双乳单纯皮下腺体切除术(保留乳头乳晕),切除左侧乳腺组织12.0 cm×8.0 cm×2.0 cm、右侧乳腺组织11.0 cm×9.0 cm×2.5 cm,术后病理切片行HE染色,光镜下见乳腺导管上皮增生及纤维脂肪组织增生(图4),符合双侧乳腺发育症的病理表现。
2017年9月11日,患者返院复诊,未诉明显不适,检查显示双侧乳房伤口愈合良好,继续予十一酸睾酮补充雄激素治疗。2017年11月,电话随访患者,患者诉双侧乳房未再触及结节,性功能基本同前,嘱其继续十一酸睾酮补充雄激素治疗。
男性乳腺发育症是最常见的男性乳腺疾病,年轻男性乳腺发育的患病率约38%[6]。男性乳腺发育症病理机制为雌激素与雄激素比例失调引起,可分为生理性及病理性男性乳腺发育症。生理性男性乳腺发育主要见于新生儿、青春发育期及健康老年患者,大部分增生的乳腺可自行缩小甚至消退[7]。长期存在的乳腺发育需注意病理性因素,如肝肾功能不全、药物、睾丸疾病、肿瘤、甲状腺疾病、性功能减退等,若发现男性性功能低下,需要完善染色体检测,排除性发育障碍疾病[1,8,9]。
XX男性综合征,亦称XX男性性反转综合征,最初由Delachapelle于1964年描述,是一种罕见的性发育障碍疾病,发病率约为男性新生儿的1/25 000~1/20 000[3]。大部分患者成年后出现不育、性功能低下等表现,42%左右患者有男性乳腺发育症[10]。约10%~20%患者儿童时因"尿道下裂、生殖器模糊、隐睾"等问题就诊[11]。激素检查提示高促性腺激素性性功能减退,染色体检测为46,XX。90%以上的XX男性综合征患者基因组中含SRY基因[12]。SRY基因常易位到X染色体上。这种46,XX (SRY+)男性的形成机制通常是由于父源生殖细胞在减数分裂时,X染色体与Y染色体之间发生不平衡交换,使带有SRY基因的Y染色体片段易位到X染色体短臂上,从而使46,XX患者的原始性腺发育为睾丸[13]。10%患者SRY基因阴性,SOX家族、RSPO1、DMRT等基因突变可能是男性性腺发育的机制之一[14,15,16]。本例患者因双侧乳腺发育入院,激素检查显示高促性腺激素性性功能减退,染色体为46,XX,SRY基因阳性,明确诊断为"XX男性综合征"。患者双侧乳腺病理检查确诊男性乳腺发育症,性激素检查示高促性腺激素性性功能减退,结合既往研究[1],考虑乳腺发育与男性性功能减退有关。患者精液检测未见精子,表现为无精子症,其原因是缺乏调控精子形成的Y染色体AZF a、b、c区域,因此,此类患者无法生育[17]。XX男性综合征占男性不育的比例为1%~9%[18]。此外,该病患者身高(163.5 cm)较其父亲(168.0 cm)及同龄正常人的平均身高低,可能原因为缺乏Y染色体长臂上的身高相关基因[19,20]。
XX男性综合征需与Klinefelter综合征鉴别:两者均有高促性腺激素性性功能减退、乳腺发育、不育等表现,但XX男性综合征患者身高较Klinefelter综合征患者低,且后者核型为46,XXY,可通过核型分析排除[21]。此外,仍需与46,XX真两性畸形鉴别,后者有卵巢和睾丸两种性腺,可通过超声检查或性腺活组织检查来鉴别。
诊治XX男性综合征的主要原则是尽早确定染色体、性腺及外生殖器性别,并矫治外生殖器畸形及相关异常。睾酮替代治疗可改善男性性功能不全,纠正男性化不足,提高生活质量[9,20]。关于此类患者乳腺发育与乳腺癌发病率的关系,暂无明确报道,但国外3篇病例报道显示XX男性综合征的乳腺发育患者发生乳腺癌[22,23,24]。雌激素水平高是乳腺癌的危险因素,因此,具有雌激素与雄激素比例失调、乳腺发育等危险因素的XX男性综合征患者应尽早行乳腺腺体切除术[1]。另外,还需关注由于雄激素低下引起骨质疏松等问题,建议此类患者完善骨密度等检查,必要时补充维生素D等改善骨质代谢[9]。此患者存在无精子症,无法生育,可通过精子库人工授精等方法孕育下一代。
综上所述,男性乳腺发育症患者应注意有无病理性因素,建议常规完善内分泌激素检查及明确染色体核型,避免漏诊性发育异常疾病。XX男性综合征可通过染色体检查及基因检测明确诊断,补充雄激素有利于纠正男性化不足。笔者建议该病乳腺发育的患者行乳腺腺体切除术。

























