
冠状动脉(简称冠脉)钙化是冠脉粥样硬化的明确证据。Agatston评分反映钙化程度和密度,其与患者发生主要急性冠脉事件的风险密切相关。动脉粥样硬化发生后,内皮细胞受损,低密度脂蛋白胆固醇(LDLc)渗透至管壁内膜下,内膜下胶原蛋白中的蛋白聚糖对LDLc具有高亲和力,将脂蛋白胆固醇复合物滞留其中。随着内皮损伤的修复,LDLc被捕获至内膜下,滞留的LDLc诱导上覆内皮的炎性反应,使内皮表达趋化肽,趋化肽吸引血液中的单核细胞,使单核细胞沿浓度梯度进入组织,成为巨噬细胞,巨噬细胞吞噬并消化动脉粥样硬化中刺激性的LDLc。在消化LDLc的过程中,巨噬细胞中的酶使LDLc复合物氧化,氧化的低密度脂蛋白(LDL)对巨噬细胞产生毒性,当其数量充足时,可导致巨噬细胞死亡,促进动脉粥样硬化的炎性反应。在坏死性炎性病灶中,调控组织中钙、磷浓度的机制失调,使得磷酸钙的溶度积过高,从而产生微小的磷酸钙晶体。随着炎性反应的持续存在,更多的磷酸钙晶体形成并聚集。当这些聚集的磷酸钙晶体超过1 mm时,就成为临床CT可识别的冠脉钙化。连续的门控心脏CT扫描证实,一旦形成CT可见的钙化,其大小不会明显减少,但可能会增加。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
尽管营养不良性血管钙化是一个有害因素,但它不能确定是否引起主要急性冠状动脉(简称冠脉)事件(major acute coronary event, MACE)。有活动性钙化的动脉粥样硬化瘤最有可能导致MACE,18F-NaF PET/CT显像可以识别这种病灶。最近数据表明,对可能引起MACE的这种病灶,18F-NaF显像是灵敏且特异的影像学标志,这一标志能否识别出高危的MACE患者需要多中心研究进一步确定。
血管钙化可发生在动脉内膜和中膜。中膜钙化多出现在外周血管,通常与钙或磷水平升高有关。钙、磷水平升高在慢性肾脏疾病和甲状旁腺功能亢进患者中常见。虽然尸检发现中膜钙化常见,但其很少侵入管腔[1]。此外,内膜钙化与动脉粥样硬化相关。动脉粥样硬化是一种病因学不明的多灶性疾病,其特征是低密度脂蛋白胆固醇(low-density lipoprotein cholesterol, LDLc)积聚在内膜下,引起称为动脉粥样硬化瘤的炎性病变。当患者血液中LDLc水平持续升高时[1],动脉粥样硬化经过数十年的进展,原有病变扩大并可形成新病变,动脉粥样硬化可形成单个或多个管腔狭窄。狭窄发生后,血液经过管壁不规则表面形成湍流,可导致血管壁局部压力升高。如果具有薄纤维帽的动脉粥样硬化瘤出现严重的炎性反应,纤维帽破裂,粥样硬化瘤中的坏死物质进入管腔[2],释放促血栓形成物质,导致血栓形成。如果血栓大到足以阻碍血流,则可能产生MACE,如心肌缺血、心肌梗死、短暂性脑缺血发作或卒中。尽管动脉粥样硬化的发生率很高,但临床事件发生率很低,基于尸检结果,产生临床事件的动脉粥样硬化破裂发生率低于1%[3]。
尽管动脉粥样硬化瘤破裂引起临床事件的发生率较低,但动脉粥样硬化导致的心血管疾病非常常见。2016年,在美国,心血管疾病占死亡总数的1/3,其中因冠脉粥样硬化性心脏病(简称冠心病)死亡360 000例[4]。尽管普遍开展了"抵制烟草、药物控制高血压和高脂血症"等活动[5],冠心病仍然是美国中年人死亡的主要原因。为了降低冠心病的死亡率,已经形成多种方法来甄别存在MACE风险的患者。
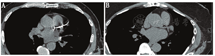
Framingham心脏研究始于20世纪50年代,已经开发了几个临床和实验室指标用来预测患者未来发生临床心血管事件的可能性。如Lee等[6]制定的修订心脏风险指数通常用作急诊外科手术患者围手术期MACE风险的初步评估(表1)。对于非急诊手术的患者,可以采用多种方法评估MACE风险,包括在线风险计算[7]和预测患者未来10年内发生MACE风险的指南。当患者存在MACE高风险时,需结合其他临床和影像学指标进一步评估,如高敏C反应蛋白检测,或用门控平扫CT[8]来检测冠脉钙化,作为评估冠心病负荷的指标(图1A和图1B)。

修订的心脏风险指数[6]
修订的心脏风险指数[6]
| 因素 | 描述 |
|---|---|
| 1 | 高风险的外科手术(较大的非心脏手术) |
| 2 | 缺血性心脏病 |
| 3 | 充血性心力衰竭病史 |
| 4 | 脑血管疾病病史 |
| 5 | 术前用胰岛素治疗的糖尿病患者 |
| 6 | 术前血清肌酐>20 mg/L |
注:具有0、1、2或3个以上因素的患者围手术期心脏并发症发生率分别为0.5%、1.3%、4%和9%
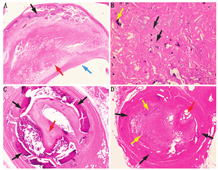
动脉粥样硬化瘤中的钙化起源于炎性的坏死核。坏死核中含有有害的氧化脂蛋白胆固醇、载脂巨噬细胞(泡沫细胞)、濒死或死亡的巨噬细胞和平滑肌细胞。动脉粥样硬化的组织病理学证实在坏死区域内含有大量直径为2~15 μm的微钙化(图2A~2D)。当高脂血症持续存在,尤其是同时存在其他心血管危险因素(如吸烟、糖尿病和不受控制的高血压)时,坏死核增大,钙化的数量和大小随之增加。在数字X线成像术出现前,心血管医师在X射线透视下,观察到冠脉钙化灶随着患者的心跳同步出现。如今,冠脉钙化可以在平扫心电门控CT上探测和量化,但CT扫描只能识别大于0.1 mm的钙化灶,CT值超过130 HU且体积至少要有3个像素才符合显著钙化的标准。尽管已经开发出几种计算钙化积分的方法[9],但最常用的是Agatston评分,其通过每支冠脉的钙化密度和钙化面积计算分支血管的钙化得分,再相加得到该患者冠脉粥样硬化负荷总得分[10]。
血管内皮除了形成半选择性屏障使大分子物质滞留在血管中,还参与了免疫反应、血管修复和生物活性分子的代谢[11]。在生理条件下,内皮细胞充当屏障,阻止血液中的LDLc到达内膜下。但是,当内皮细胞因外伤、感染、炎性反应或自身免疫而受损时,损伤的内皮组织通透性增加,LDLc可以穿过血管内皮,脂蛋白胆固醇复合物和内膜下富含胶原蛋白的蛋白聚糖接触,并被滞留在内膜下。随着相邻的内皮细胞修复该处内皮损伤,内膜下的LDLc被捕获,LDLc中的胆固醇可形成结晶[12],引起病灶体积膨胀和局部炎性反应[13]。炎性的内皮细胞表达趋化肽[14,15],吸引血液中的单核细胞进入内皮下组织。浸润的单核细胞分化为巨噬细胞以吞噬脂蛋白-脂质复合物。在吞噬和消化脂蛋白结合胆固醇的过程中,巨噬细胞产生氧化LDLc复合物的酶。氧化的LDLc对巨噬细胞产生毒性,当数量充足时,会导致巨噬细胞死亡。动脉粥样硬化中的其他细胞,如平滑肌细胞,也可能在强烈的炎性反应环境中死亡。
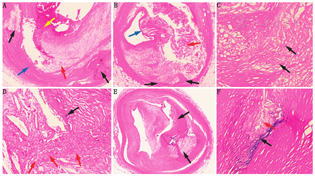
早期动脉粥样硬化仅表现为低水平的炎性反应。在这一阶段,巨噬细胞的死亡通常是由细胞凋亡等程序化死亡引起[16]。当细胞因凋亡而死亡时,死亡细胞的残屑被邻近细胞吞噬,从而使病灶处的残留炎性反应最小化。在较大、严重的炎性病灶中,细胞死亡方式不仅只有凋亡,还有无序化死亡,如坏死性凋亡和细胞坏死。后者可使死亡细胞的残屑滞留在病灶中,从而加重炎性反应。这种低效清除死亡细胞残屑的模式称为胞葬缺陷[17],死亡和濒死细胞的残屑增加,并至动脉粥样硬化瘤的坏死核,从而使病灶扩大、炎性反应增强,形成营养不良性钙化(图3)[18,19]。
目前,服用降脂药物[如他汀类药物和前白蛋白转化酶枯草菌素9(proprotein convertase subtilisin/Kexin type 9, PCSK9)抑制剂]治疗高脂血症是近年来的标准治疗方案。除了药物治疗之外,生活方式的改变可以减轻动脉粥样硬化的炎性反应,包括运动、保持正常的体质指数、糖尿病患者有效控制血糖以及地中海式饮食。但如果患者继续追求致动脉粥样硬化的生活方式,动脉粥样硬化中的炎性反应可能会持续存在或加剧。
在健康细胞中,钙[20]和磷[21]的局部浓度受到严格调控,以最大程度抑制有害磷酸钙晶体形成的可能。另一方面,在炎性动脉粥样硬化坏死核中濒死和坏死细胞的残屑,不受钙和磷酸盐浓度调节机制的调控,使细胞中的这些离子超过溶度积,形成磷酸钙晶体。如碱性磷酸酶、腺苷三磷酸酶和坏死核的活性氧自由基,会通过分解大分子物质而产生游离的磷酸盐[22]。在炎性反应、凋亡、坏死性细胞崩解和细胞质膜出芽[23]过程中产生的微泡[24,25,26]可以作为磷酸钙晶体形成的支架[27]。根据提供微泡的细胞,颗粒可能含有促进磷酸钙晶体形成的物质或抑制血管钙化的物质。当坏死核中形成磷酸钙晶体时,它们也可能捕获其他离子,如镁。
平扫门控CT扫描可以定量提供同时反映冠脉钙化程度和密度的值[10,28,29,30]。最初在坏死核中形成约2~15 μm的晶体太小,在空间分辨率约为0.6 mm的CT上无法识别(图2)。随着时间的推移,在持续高脂血症的存在下,坏死核中会形成新的微晶体并聚集起来。30年前使用电子束CT和最近使用平扫心电门控CT的研究表明,Agatston评分较高的患者发生MACE的风险更大[31]。然而,最近有研究表明,冠脉钙化的体积和密度对预后有不同的影响[32]。较高密度钙化可能反映较低的脂质含量或先前亚临床斑块破裂愈合,提示重度钙化的病灶稳定[33],预后较好。相反,多发小灶的低密度钙化,特别是病灶位于脂质池区域(如斑点状或碎片状钙化),表明病灶不稳定,发生MACE的风险较高。
几个大型试验,如PROSPECT(冠脉树局部观察预测事件研究)子研究[34],也发现罪犯病灶的钙化密度较稳定病灶低,这支持了高密度钙化与病变稳定相关[35]。
可以通过分析动脉粥样硬化斑块的机械应力评估其稳定性[36]。与单一病灶存在时相比,当微钙化与坏死核中的脂质在动脉粥样硬化瘤表面共存时,该区域管壁应力面积增加[37]。其他影响动脉粥样硬化帽应力的因素包括微钙化的位置(在病灶近表面或病灶深处)、微钙化的大小、单个钙化还是多灶钙化、病灶方向与血流方向是否一致。
动脉粥样硬化中的大多数微钙化发生在死亡或濒死的巨噬细胞附近,位于坏死脂核的深处,很少在纤维帽中(如果在纤维帽中,应力增加易导致斑块破裂)。
使用实验室专用高分辨率CT扫描仪对人冠脉标本中钙化进行检测,发现62个冠脉粥样硬化瘤中有数千个微钙化(其中97%小于0.2 mm),而9个冠脉粥样硬化的纤维帽中仅有81个微钙化灶[38]。三维有限元分析表明,如果动脉粥样硬化瘤纤维帽中存在2个微钙化点灶,彼此分离,且病灶方向与血流拉伸轴方向一致,则粥样硬化纤维帽的周向应力峰值会增加5倍,这种水平的应力足以使纤维帽破裂。另一方面,当微钙化位于黏性的坏死核中,并且与纤维帽分离,则钙化灶不增加粥样硬化帽上的剪切应力[39]。
肉眼可见的冠脉钙化不可逆,它们是由于失控的、不适当的刺激造成病理过程的最终结果[40]。钙化是主动还是被动所致尚不清楚[41]。
连续的CT测量可显示钙化要么是稳定的,要么随时间增加[42,43]。尽管一些研究认为某些病灶的钙化轻度减少,但这些所谓的轻度减少可能是由于技术因素引起,而不是该部位钙化数量的真正减少[42]。一项长期动物研究也支持钙化持续存在,在该研究中,首先给猴子喂食致动脉粥样硬化的食物,然后延长喂食使动脉粥样硬化消退的食物[44]。对在致动脉粥样硬化饮食阶段处死的猴冠脉进行组织病理学分析,结果表明细胞内和细胞外的钙颗粒通常与大脂质池相邻。进行动脉粥样硬化消退饮食后3.5年,对处死的猴冠脉进行组织病理学分析,结果表明在没有脂质池的情况下仍有局部钙化,支持了钙化不会减少或消退的结论。
在生理条件下,多种因素可抑制或预防[45,46]肉眼可见的血管钙化。
1.胎球蛋白A(59×103糖蛋白)。这种糖蛋白由肝脏产生,可与小的磷酸钙晶体结合,形成被巨噬细胞吞噬的钙蛋白颗粒。
2.骨钙蛋白。成骨细胞产生的这种维生素K依赖的基质蛋白可抑制磷灰石晶体的生长。
3.基质γ-羧基谷氨酸蛋白(matrix γ-carboxyglutamate Gla protein, MGP)。该14×103蛋白可吸收结晶磷酸钙。骨钙蛋白和MGP有赖于维生素K激活[47]。MGP由血管平滑肌细胞、软骨细胞、内皮细胞和成纤维细胞合成,并在局部分泌。MGP通过螯合钙、磷酸盐离子以及晶体与磷酸钙结合[46]。当缺乏维生素K或维生素K含量降低时,MGP失活,依赖维生素K的凝血因子Ⅱ、Ⅶ、Ⅸ和Ⅹ也同样失活[48,49]。维生素K拮抗剂华法林(香豆素;美国百时美施贵宝公司)通过抑制维生素K环氧化物还原酶来抑制维生素K活化。在缺乏活性维生素K的情况下,这些限制血管钙化的重要物质失活,导致接受香豆素治疗的患者出现令人惊讶的血管钙化增加。一项由66例患者组成的随机试验比较了使用华法林与阿哌沙班治疗后心房颤动患者冠脉钙化的变化[50],使用华法林治疗的患者钙化和低衰减斑块体积高于阿哌沙班治疗组。同样,对8项使用血管内超声的前瞻性随机试验的分析表明,与未使用华法林的患者相比,使用华法林的患者的年钙化指数显著增加[51]。
老化会导致传导动脉的僵硬度和动脉粥样硬化增加[52,53]。衰老的发生,部分是因为成体干细胞不能永生。Hayflick和Moorhead[54]证明,正常人的二倍体细胞在体外复制能力有限(约40代),在其复制的末期,最后一代细胞丧失复制力。这种复制力受限的部分原因可能是每次分裂都会减少每个分区的端粒长度。随着年龄的增长,传导血管中的胶原蛋白逐渐增加,弹性蛋白逐渐减少。弹性蛋白下降的原因是由于每次心跳时血管的反复扩张和收缩而导致的"疲劳衰竭"[55]。在没有吸烟、高血压、高脂血症或葡萄糖耐量异常等危险因素的情况下,年龄与冠心病风险增加相关[56]。老龄还与"血管扩张、管壁增厚、细胞和细胞外基质重组、胶原和单核细胞聚集造成的血管内膜下间隙增加有关"[56]。细胞外基质富含糖胺聚糖,可滞留LDLc,而LDLc可轻易穿透衰老的内皮细胞[57,58,59]。
用他汀类药物治疗可降低血液中LDLc水平但增加冠脉血管钙化,使动脉粥样硬化趋于稳定[60]。血液中较低水平的LDLc可有效减少内膜下脂质聚集,减少炎性反应,缩小坏死核,同时可增加坏死核的钙化。
识别有猝死风险的患者仍是一个重大挑战[5]。2014年发布了一篇评论[61],总结了多种影像技术与MACE的关系,包括有创血管内超声、光学相干断层扫描以及增强CT等,结果表明影像检测对MACE的阳性预测值为4%~22%,其结果不足以指导无症状患者(即使有比较低的风险会发生不良事件)进行经皮介入治疗(如支架植入)。期待有更高阳性预测值的影像检测技术指导无症状患者的治疗[62]。
一些分子影像技术可以对动脉粥样硬化瘤定位和特征诊断[62],包括放射性标记的自体LDLc[63]、放射性标记可识别氧化LDLc的抗体[64]、放射性标记的生长抑素受体拮抗剂(炎性动脉粥样硬化瘤表达的生长抑素受体显像)[65]、放射性标记的动脉粥样硬化乏氧的定位显像[66]、局灶性血管炎性反应的18F-脱氧葡萄糖(fluorodeoxyglucose, FDG)显像[67]以及近期应用的可识别活动性钙化的18F-NaF显像[68]。尽管18F-FDG定位并聚集在有症状的颈动脉疾病患者的颈动脉区域,但在与MACE相关的冠脉病变处18F-FDG摄取是否增高仍不清楚[69]。在急性心肌梗死和稳定型心绞痛患者中,18F-NaF与18F-FDG相比,罪犯斑块摄取NaF明显增高[70]。已经证实18F-NaF显像探测高危斑块病灶有较高的灵敏度,但这些通过光学相干断层扫描、CT和血管内超声判断为高危的斑块病灶并不摄取18F-FDG[71]。动脉粥样硬化病灶处18F-NaF的摄取与光学相干断层扫描中所见病灶处存在的微血管相关,提示18F-NaF病灶处的摄取与炎性反应相关。目前认为18F-NaF显像对于无临床症状但发生MACE风险较高且需要治疗的患者具有较高的阳性预测值,但该结论尚需进行前瞻性多中心研究进一步验证。
平扫CT所见的冠脉钙化灶表明冠脉粥样硬化的存在。CT表现为动脉粥样硬化内存在点状钙化且邻近脂质池,提示斑块不稳定;另一方面,如果钙化致密,呈板状,特别是在远离脂质池的情况下,提示病灶稳定。18F-NaF PET/CT显像可在一次显像中检测出多个血管区域的活动性钙化灶,据此通过全身扫描可识别多个血管区域的高危病灶。
感谢Heiko Schoder医学博士的编辑建议及其对图1的提供与数据临床描述
























