探讨白细胞介素17A(IL-17A)调控小鼠角质形成细胞(KC)表达IL-1β和IL-23的机制。
从400只新生雌雄不限C57BL/6野生型小鼠皮肤中分离原代KC,用含体积分数10%胎牛血清的RPMI 1640培养基培养于24孔板中,用于以下实验。(1)取细胞,采用随机数字表法(分组方法下同)分为磷酸盐缓冲液(PBS)对照组、IL-17A刺激组,分别加入10 μL的PBS、质量浓度为100 ng/mL的IL-17A培养6 h,采用实时荧光定量反转录PCR法检测细胞中IL-1β和IL-23 mRNA表达水平,每组3个样本。(2)取细胞,分为二甲基亚砜(DMSO)对照组、IL-17A+DMSO组、IL-17A+核因子κB抑制剂组、IL-17A+信号转导及转录激活因子3(STAT3)抑制剂组、IL-17A+胞外信号调节激酶1(ERK1)抑制剂组、IL-17A+ERK2抑制剂组、IL-17A+c-Jun氨基端激酶(JNK)抑制剂组,分别加入相应试剂,各试剂体积均为10 μL,IL-17A质量浓度为100 ng/mL,核因子κB、STAT3、ERK1、ERK2、JNK信号通路抑制剂PDTC、S3I-201、SCH772984、SCH772984、SP600125物质的量浓度分别为5 μmol/L、100 μmol/L、4 nmol/L、1 nmol/L、10 μmol/L,均培养6 h。采用实时荧光定量反转录PCR法检测细胞中IL-1β和IL-23 mRNA表达水平,每组3个样本。(3)取细胞,同实验(1)分组处理,采用蛋白质印迹法检测细胞中核因子κB磷酸化、STAT3磷酸化、ERK磷酸化、JNK磷酸化水平,每组3个样本。对数据行双尾Student t检验、单因素方差分析、t检验和Bonferroni校正。
(1)培养6 h,与PBS对照组比较,IL-17A刺激组细胞中IL-1β和IL-23 mRNA表达水平均明显升高(t=13.46、6.72,P<0.01)。(2)培养6 h,DMSO对照组、IL-17A+DMSO组、IL-17A+核因子κB抑制剂组、IL-17A+STAT3抑制剂组、IL-17A+ERK1抑制剂组、IL-17A+ERK2抑制剂组、IL-17A+JNK抑制剂组细胞中IL-1β与IL-23 mRNA表达水平分别为1.00±0.11、4.01±0.32、0.32±0.06、1.76±0.43、3.62±0.24、3.80±0.43、4.26±0.74和1.03±0.29、4.08±0.34、4.76±0.38、4.70±0.21、1.06±0.42、0.92±0.21、0.39±0.05。与DMSO对照组比较,IL-17A+DMSO组细胞中IL-1β和IL-23 mRNA表达水平均明显升高(t=9.24、12.60,P<0.01)。与IL-17A+DMSO组比较,IL-17A+核因子κB抑制剂组与IL-17A+STAT3抑制剂组细胞中IL-1β mRNA表达水平均明显下降(t=11.34、6.91,P<0.01),IL-17A+ERK1抑制剂组、IL-17A+ERK2抑制剂组和IL-17A+JNK抑制剂组细胞中IL-23 mRNA表达水平均明显下降(t=12.44、13.03、15.21,P<0.01)。(3)培养6 h,与PBS对照组比较,IL-17A刺激组细胞中核因子κB磷酸化、STAT3磷酸化、ERK磷酸化、JNK磷酸化水平均明显升高。
IL-17A分别通过促进核因子κB、STAT3信号通路磷酸化与ERK、JNK信号通路磷酸化促进小鼠KC转录表达IL-1β与IL-23。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
IL-17A是促炎性细胞因子,可招募中性粒细胞和巨噬细胞,在炎症反应的起始和发展中发挥着重要作用[1,2,3,4]。在皮肤移植和创面愈合中,Vγ4 T细胞是创缘组织IL-17A的早期主要来源,可显著促进移植排斥反应和抑制创面愈合[5,6]。IL-1β和IL-23对于炎症反应的启动和放大非常重要[7,8,9,10,11]。皮肤中的KC和朗格汉斯细胞是IL-1β和IL-23的主要来源[12,13]。在接触过敏性皮炎、银屑病样皮炎、抗原诱导关节炎中,IL-1β和IL-23可刺激γδT细胞产生IL-17A[12,13,14,15]。本课题组观察到IL-1β和IL-23可诱导Vγ4 T细胞产生IL-17A,从而加重移植排斥反应和创面愈合延迟[5,6]。有意思的是,Vγ4 T细胞产生的IL-17A可反过来增强KC中IL-1β和IL-23的表达,即IL-1β/IL-23与IL-17A构成一个相互促进的正反馈回路[5,6]。然而Vγ4 T细胞分泌的IL-17A调控KC表达IL-1β和IL-23的机制尚未明确。
IL-17A发挥作用的信号通路众多,其中研究较为明确的有胞外信号调节激酶1/2(ERK1/2)、c-Jun氨基端激酶(JNK)、信号转导及转录激活因子3(STAT3)、核因子κB等信号通路[16,17,18,19,20,21,22,23,24,25]。在本研究中,这几个通路的抑制剂被选中进行细胞实验,观察其能否阻断IL-17A对KC的刺激表达作用[19,26,27,28,29,30,31]。本课题重点探讨IL-17A影响IL-1β和IL-23在KC中表达的机制,以期对创面愈合机制进行有益补充。
本研究遵循陆军军医大学和国家有关实验动物管理和使用的规定。
400只健康无特殊病原体级雌雄不限C57BL/6野生型新生小鼠购自陆军军医大学实验动物中心,许可证号:SCXK(军)2017-0024。CD11c超纯微磁珠分选试剂购自德国美天旎生物技术有限公司,分散酶Ⅱ粉末购自德国Sigma公司,胎牛血清、胰蛋白酶购自美国Gibco公司,RPMI 1640培养基、二甲基亚砜(DMSO)购自北京索莱宝科技有限公司,PBS购自武汉博士德生物工程有限公司,IL-17A购自美国R&D公司,核因子κB信号通路抑制剂PDTC、STAT3信号通路抑制剂S3I-201、ERK信号通路抑制剂SCH772984、JNK信号通路抑制剂SP600125购自美国Selleck生物科技有限公司,Qiagen RNeasy试剂盒购自美国Invitrogen生命技术公司,TaqMan反转录试剂盒购自美国Life Technologies公司,IL-1β和IL-23引物由生工生物工程(上海)股份有限公司设计、合成,SYBR Green实时荧光定量试剂盒购自日本TaKaRa公司,裂解缓冲液与酶抑制剂购自江苏凯基生物技术股份有限公司,兔抗小鼠核因子κB p65、磷酸化核因子κB p65、STAT3、磷酸化STAT3、ERK1/2、磷酸化ERK1/2、JNK、磷酸化JNK单克隆抗体购自美国Cell Signaling Technology公司,辣根过氧化物酶(HRP)标记的山羊抗兔IgG抗体、兔抗小鼠GAPDH单克隆抗体购自天津三箭生物技术股份有限公司。凝胶电泳仪、ChemiDoc™ XRS检测系统购自美国Bio-Rad公司,StepOne Plus型实时荧光定量PCR仪购自美国Applied Biosystems公司,FACSCantoⅡBD型流式细胞仪购自美国BD公司,710型多功能酶标仪购自美国BioTek仪器有限公司。
所有动物实验均在无特殊病原体条件下进行,每次取20只新生小鼠(本研究共用400只),用10 g/L戊巴比妥钠溶液以50 mg/kg腹腔注射麻醉,脱颈处死并剥离皮肤。刮除皮肤皮下筋膜组织,将皮肤修剪为1.0 cm×0.5 cm大小的皮片。将皮片表皮面朝上、真皮面朝下悬浮在5 mg/mL的分散酶Ⅱ溶液中,在体积分数5%二氧化碳、37 ℃孵箱中孵育1~2 h。剥下表皮层并充分剪碎,加入适量3 g/L胰蛋白酶溶液并充分吹打混匀,放入体积分数5%二氧化碳、37 ℃孵箱中孵育5~10 min。加入4倍体积的含体积分数10%胎牛血清的RPMI 1640培养基中和胰蛋白酶溶液,并轻柔吹打混匀。将混悬液经滤孔孔径为70 μm滤器过滤至15 mL离心管中,于室温下,以800×g离心8 min。弃去上清液,用含体积分数10%胎牛血清的RPMI 1640培养基重新悬浮细胞,将细胞浓度调整为5×105~1×106个/mL。按照CD11c超纯微磁珠分选试剂说明书上的步骤,将CD11c+细胞从原代KC中分选弃除,分离后的KC纯度大于95%,后续实验使用原代细胞。
采用实时荧光定量RT-PCR法检测。将KC浓度调整为1×106个/mL,接种至24孔板中,每孔1 mL(细胞浓度和接种方法下同)。按照随机数字表法(分组方法下同),将细胞分为PBS对照组、IL-17A刺激组(每组3孔),分别加入10 μL的PBS、质量浓度为100 ng/mL的IL-17A培养6 h。使用Qiagen RNeasy试剂盒提取细胞的总RNA,检测RNA纯度及浓度。取1 μg RNA稀释,使用TaqMan反转录试剂盒按20 μL反应体系将RNA反转录为互补DNA。使用实时荧光定量PCR仪检测IL-1β和IL-23的mRNA表达。IL-1β上游引物为5′-ACCTTCCAGGATGAGGACATGA-3′、下游引物为5′-CTAATGGGAACGTCACACACCA-3′,产物大小为116 bp;IL-23上游引物为5′-CTGAGCCACCCAGGAAAGTA-3′、下游引物为5′-TGAGAAAACCCAGAGCATCA-3′,产物大小为101 bp;GAPDH上游引物为5′-CGTGCCGCCTGGAGAAAC-3′、下游引物为5′-AGTGGGAGTTGCTGTTGAAGTC-3′,产物大小为138 bp。基因表达水平以GAPDH表达为内参照。PCR条件:95 ℃预变性10 min;95 ℃变性5 s,60 ℃退火34 s,共40个循环。采用Δ循环阈值(Ct)法计算目的基因的相对表达水平,即2-ΔΔCt。本实验重复3次,取最具代表性的1次结果进行展示。
采用实时荧光定量RT-PCR法检测。将KC分为DMSO对照组、IL-17A+DMSO组、IL-17A+核因子κB抑制剂组、IL-17A+STAT3抑制剂组、IL-17A+ERK1抑制剂组、IL-17A+ERK2抑制剂组、IL-17A+JNK抑制剂组(每组3孔),分别加入10 μL DMSO、10 μL质量浓度为100 ng/mL的IL-17A+10 μL DMSO、10 μL质量浓度为100 ng/mL的IL-17A+10 μL物质的量浓度为5 μmol/L的PDTC、10 μL质量浓度为100 ng/mL的IL-17A+10 μL物质的量浓度为100 μmol/L的S3I-201、10 μL质量浓度为100 ng/mL的IL-17A+10 μL物质的量浓度为4 nmol/L的SCH772984、10 μL质量浓度为100 ng/mL的IL-17A+10 μL物质的量浓度为1 nmol/L的SCH772984、10 μL质量浓度为100 ng/mL的IL-17A+10 μL物质的量浓度为10 μmol/L的SP600125,均培养6 h。同1.3进行IL-1β和IL-23 mRNA检测,本实验重复3次,取最具代表性的1次结果进行展示。
采用蛋白质印迹法检测。将KC同1.3进行分组处理,每组3孔。收集各组KC,加入现配的裂解缓冲液与酶抑制剂混合溶液,冰上提取其胞内蛋白质,测定蛋白浓度,加热变性。每孔取30 μg蛋白,行十二烷基硫酸钠-聚丙烯酰胺凝胶电泳,湿法转膜。3 g/L牛血清白蛋白溶液室温摇动封闭2 h,分别加入兔抗小鼠核因子κB p65、磷酸化核因子κB p65、STAT3、磷酸化STAT3、JNK、磷酸化JNK、ERK1/2、磷酸化ERK1/2、GAPDH单克隆一抗(稀释比均为1∶1 000),4 ℃孵育过夜。加入HRP标记的山羊抗兔IgG二抗(稀释比为1∶5 000),摇床室温孵育1 h。将超敏发光液A、B根据体积以1∶1比例现配现用,将膜浸泡于化学发光剂不超过30 s,转移至ChemiDoc™ XRS检测系统中读取核因子κB p65、磷酸化核因子κB p65、STAT3、磷酸化STAT3、JNK、磷酸化JNK、ERK1/2、磷酸化ERK1/2、GAPDH表达图像。本实验重复3次,取最具代表性的1次结果进行展示。
采用SPSS 19.0统计软件进行分析数据,计量资料数据均符合正态分布,以 ±s表示。2组分组组间比较行双尾Studentt检验;多组分组组间总体比较行单因素方差分析,组间两两比较行t检验并进行Bonferroni校正。P<0.05为差异有统计学意义。
±s表示。2组分组组间比较行双尾Studentt检验;多组分组组间总体比较行单因素方差分析,组间两两比较行t检验并进行Bonferroni校正。P<0.05为差异有统计学意义。
培养6 h,与PBS对照组比较,IL-17A刺激组细胞中IL-1β和IL-23 mRNA表达水平均明显升高(P<0.01),见表1。
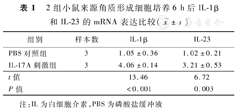
2组小鼠来源角质形成细胞培养6 h后IL-1β和IL-23的mRNA表达比较( ±s)
±s)
2组小鼠来源角质形成细胞培养6 h后IL-1β和IL-23的mRNA表达比较(±s)
| 组别 | 样本数 | IL-1β | IL-23 |
|---|---|---|---|
| PBS对照组 | 3 | 1.05±0.36 | 1.02±0.21 |
| IL-17A刺激组 | 3 | 4.06±0.14 | 3.21±0.53 |
| t值 | 13.46 | 6.72 | |
| P值 | <0.001 | 0.003 |
注:IL为白细胞介素,PBS为磷酸盐缓冲液
培养6 h,与DMSO对照组比较,IL-17A+DMSO组细胞中IL-1β和IL-23 mRNA表达水平均明显升高(P<0.01);与IL-17A+DMSO组比较,IL-17A+核因子κB抑制剂组与IL-17A+STAT3抑制剂组细胞中IL-1β mRNA表达水平均明显下降(P<0.01),IL-17A+ERK1抑制剂组、IL-17A+ERK2抑制剂组和IL-17A+JNK抑制剂组细胞中IL-1β mRNA表达水平无明显变化(P>0.05);与IL-17A+DMSO组比较,IL-17A+核因子κB抑制剂组与IL-17A+STAT3抑制剂组细胞中IL-23 mRNA表达水平无明显变化(P>0.05),IL-17A+ERK1抑制剂组、IL-17A+ERK2抑制剂组和IL-17A+JNK抑制剂组细胞中IL-23 mRNA表达水平均明显下降(P<0.01)。见表2。
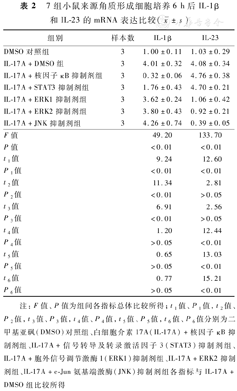
7组小鼠来源角质形成细胞培养6 h后IL-1β和IL-23的mRNA表达比较( ±s)
±s)
7组小鼠来源角质形成细胞培养6 h后IL-1β和IL-23的mRNA表达比较(±s)
| 组别 | 样本数 | IL-1β | IL-23 |
|---|---|---|---|
| DMSO对照组 | 3 | 1.00±0.11 | 1.03±0.29 |
| IL-17A+DMSO组 | 3 | 4.01±0.32 | 4.08±0.34 |
| IL-17A+核因子κB抑制剂组 | 3 | 0.32±0.06 | 4.76±0.38 |
| IL-17A+STAT3抑制剂组 | 3 | 1.76±0.43 | 4.70±0.21 |
| IL-17A+ERK1抑制剂组 | 3 | 3.62±0.24 | 1.06±0.42 |
| IL-17A+ERK2抑制剂组 | 3 | 3.80±0.43 | 0.92±0.21 |
| IL-17A+JNK抑制剂组 | 3 | 4.26±0.74 | 0.39±0.05 |
| F值 | 49.20 | 133.70 | |
| P值 | <0.01 | <0.01 | |
| t1值 | 9.24 | 12.60 | |
| P1值 | <0.01 | <0.01 | |
| t2值 | 11.34 | 2.81 | |
| P2值 | <0.01 | >0.05 | |
| t3值 | 6.91 | 2.56 | |
| P3值 | <0.01 | >0.05 | |
| t4值 | 1.20 | 12.44 | |
| P4值 | >0.05 | <0.01 | |
| t5值 | 0.65 | 13.03 | |
| P5值 | >0.05 | <0.01 | |
| t6值 | 0.77 | 15.21 | |
| P6值 | >0.05 | <0.01 |
注:F值、P值为组间各指标总体比较所得;t1值、P1值,t2值、P2值,t3值、P3值,t4值、P4值,t5值、P5值,t6值、P6值分别为二甲基亚砜(DMSO)对照组、白细胞介素17A(IL-17A)+核因子κB抑制剂组、IL-17A+信号转导及转录激活因子3(STAT3)抑制剂组、IL-17A+胞外信号调节激酶1(ERK1)抑制剂组、IL-17A+ERK2抑制剂组、IL-17A+c-Jun氨基端激酶(JNK)抑制剂组各指标与IL-17A+DMSO组比较所得
培养6 h,与PBS对照组比较,IL-17A刺激组细胞中核因子κB p65磷酸化水平明显升高(图1A);STAT3磷酸化位点705磷酸化水平明显升高(图1B),而STAT3磷酸化位点727磷酸化水平没有明显改变(图1C);ERK磷酸化水平明显升高(图1D);JNK磷酸化水平明显升高(图1E)。

注:GAPDH为3-磷酸甘油醛脱氢酶;1.磷酸盐缓冲液对照组,2.白细胞介素17A刺激组
γδ T细胞是皮肤免疫防御的重要组成部分,参与银屑病、肿瘤、皮肤移植排斥、微生物感染和创面愈合等疾病或生理过程[1,5,32,33,34,35]。γδ T细胞有不同亚群分布于皮肤的表皮和真皮层,表皮层主要为Vγ5 T细胞,因其具有树突状外形故又被称为树突状表皮T细胞(DETC);真皮层主要为Vγ4 T细胞[36,37]。皮肤损伤后创缘的DETC可快速活化分泌胰岛素样生长因子Ⅰ(IGF-Ⅰ),是表皮组织IGF-Ⅰ的主要来源,可显著促进KC增殖和迁移,对于及时有效的创面愈合而言至关重要[38]。而本课题组在前期研究中观察到,Vγ4 T细胞可阻碍DETC表达IGF-Ⅰ,进而抑制创面愈合[6]。深入探究此机制有望为创面快速愈合提供理论依据和新的治疗靶点。本课题组通过进一步探究了解到,Vγ4 T细胞分泌的IL-17A促进表皮KC表达IL-1β和IL-23,继而抑制DETC表达IGF-Ⅰ[6]。IL-1β和IL-23抑制DETC表达IGF-Ⅰ的机制为活化核因子κB和STAT3信号通路[6]。然而IL-17A促进表皮KC表达IL-1β和IL-23的机制尚不清楚,故而本课题组设计实验以进行探索。
IL-17A导致局部炎症反应放大的重要机制是刺激局部细胞产生促炎细胞因子[39]。本课题组行体外实验观察到IL-17A可直接刺激KC高表达IL-1β mRNA,而核因子κB抑制剂加入后可显著抑制KC表达IL-1β mRNA。该实验结果提示IL-17A有可能通过激活核因子κB促进IL-1β转录表达。那么IL-17A是否影响核因子κB的磷酸化水平呢?对此,本研究进行磷酸化蛋白表达检测,结果证实IL-17A处理后的KC核因子κB的磷酸化水平明显提高。上述实验结果提示,IL-17A可能通过促进核因子κB的磷酸化从而提高IL-1β mRNA的转录水平。此外,文献报道STAT3信号通路对IL-17A表达至关重要[40],如在银屑病中IL-17A可促进KC的STAT3磷酸化和核转位[18]。因此,本课题组就STAT3抑制剂对IL-17A促进KC表达IL-1β mRNA的影响和IL-17A对STAT3磷酸化的影响分别进行检测。实验结果证实STAT3抑制剂也可阻碍IL-17A对KC内IL-1β转录表达的促进作用,而IL-17A可提高STAT3磷酸化位点705的磷酸化水平。该结果提示,IL-17A可能通过促进STAT3的磷酸化从而提高IL-1β mRNA的转录水平。
此外,体外实验结果提示IL-17A也可直接刺激KC高表达IL-23 mRNA。因此本课题组检测核因子κB、STAT3、ERK1/2、JNK信号通路抑制对IL-17A刺激KC表达IL-23 mRNA的影响。然而结果提示IL-17A促进IL-1β和IL-23表达的通路各不相同。实时荧光定量RT-PCR检测到ERK1抑制剂、ERK2抑制剂以及JNK抑制剂可显著抑制IL-17A对KC表达IL-23 mRNA的促进作用,那么IL-17A是否可以促进ERK和JNK磷酸化值得探究。因为ERK1和ERK2信号通路抑制均可阻碍IL-23的表达,故在蛋白磷酸化实验中,本课题设计检测总ERK的变化。蛋白质印迹法结果证实,IL-17A刺激组KC的ERK和JNK磷酸化水平升高。这些结果提示IL-17A可能通过促进ERK和JNK磷酸化促进IL-23 mRNA的表达。
综上所述,IL-17A可能通过活化KC的核因子κB和STAT3信号通路促进IL-1β转录表达,或活化ERK和JNK信号通路促进IL-23的转录表达,后续实验需进一步探究这些信号通路是否以及如何对IL-1β或IL-23的表达调控作用起协同作用。如何有效地干预核因子κB和STAT3或ERK与JNK信号通路进而阻碍IL-17A对IL-1β或IL-23的刺激表达作用,可为创面愈合的治疗提供新思路。
所有作者均声明不存在利益冲突


























