Turner综合征又称为先天性卵巢发育不全综合征,是由于X染色体畸变导致女童生长发育异常的一种常见疾病,临床发病率为活产女婴1/5000~1/2000。X染色体畸变除导致患儿矮小、第二性征不发育和原发性闭经外,还可引起许多其他系统的疾病或异常,如心血管或肾脏畸形,听力或脊柱异常等。现就近年来Turner综合征的诊断(包括产前诊断、婴儿期诊断),定期临床随访项目、以矮小和青春期发育异常为重点的治疗等最新进展作一介绍。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
先天性卵巢发育不全综合征,只发生在女童,发病率为活产女婴的1/5000~1/2000,由性染色体畸变所致。患者的卵巢组织退化萎缩成条束状,故缺乏雌性激素,导致第二性征不发育和原发性闭经。此外还有矮小、躯体畸形等多种临床表现。该综合征1938年由美国医师Turner[1]首先报道,故也称为Turner综合征(Turner syndrome,TS)。现以矮小和青春期发育异常的临床处理为重点介绍近年来TS的诊治进展。
TS的发生是由于在细胞减数分裂或有丝分裂时,完全或部分丢失1条X染色体。在所有的胎儿中TS的发生率约占3%,自然流产的胎儿中TS约占10%,约99%核型为45,XO的胎儿在母亲怀孕的早期或中期自然流产[1]。XO/XX嵌合体的胎儿易成活,病情较轻。XO细胞比例越高,畸形相对较多;XO细胞比例越低,则畸形相对越少。临床上核型为45,XO的TS最为常见,其他核型(如嵌合体或X染色体结构异常)及其临床发生率见表1[2]。
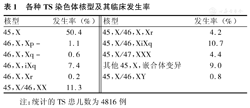
各种TS染色体核型及其临床发生率
各种TS染色体核型及其临床发生率
| 核型 | 发生率(%) | 核型 | 发生率(%) |
|---|---|---|---|
| 45,X | 50.4 | 45,X/46,X,Xr | 4.2 |
| 46,X,Xp- | 1.1 | 45,X/46,XiXq | 10.7 |
| 46,X,Xq- | 0.6 | 45,X/47,XXX | 4.4 |
| 46,X,iXq | 7.4 | 其他45,X,嵌合体变异 | 9.0 |
| 46,X,Xr | 0.2 | 45,X/46,XY | 0.8 |
| 45,X/46,XX | 11.3 |
注:统计的TS患儿数为4816例
多数TS患儿矮小,宫内轻度生长落后,出生时身长短(-0.7 SDS)、体质量低,部分为小于胎龄(SGA)儿,1~2岁生长缓慢(-1.6~1.8 SDS),3~4岁后可达矮小(-2.0 SDS)[3];无青春期生长加速,约14岁身高落后最明显。英国[4]、日本[5]、中国[6]等国家于1985年、1992年和2001年分别绘制了各自的TS患儿的自然生长曲线图,这3个国家未治疗的TS的成年平均身高分别为142.9 cm、141.2 cm和140.0 cm。据估计矮小女童中1/100~1/50为TS[7]。
过去认为TS矮小的原因:(1)生长激素(GH)分泌不足,内源性GH分泌受损,GH激发试验不能反映GH的异常;(2)GH抵抗,生理剂量GH治疗常无效,但高剂量可促进生长。新近认为与矮小同源盒基因(SHOX,short-stature homeobox-containing gene)及其他Xp上的基因单倍体剂量不足有关[8,9]。健康女性有2条X染色体,每一条X染色体上的SHOX基因均有功能-抑制生长板的融合,这是维持正常身高所必需。TS患儿缺一条X染色体或X染色体其他类型的畸变,只有一条X有功能,导致SHOX基因表达不足,使远端肢体生长板提前融合[10]。
SHOX基因缺失或突变临床表型为矮小,伴或不伴躯体畸形:远端肢体短(前臂和小腿),肘外翻,第4掌骨短,高腭穹等[11]。前期研究发现,与健康对照儿童相比TS患儿骨龄落后0.09~5.50(2.56±1.78)岁,青春期前以腕骨的成骨作用受损为著,在青春期则以长骨生长过程受损为主[12]。此外,骨龄片还显示尺骨、桡骨干骺端距离增大等骨骼畸形特征。
TS患儿外生殖器为婴儿型,小阴唇发育不良,子宫不能触及。由于X染色体异常,多数卵巢发育不全,甚至呈纤维条束状萎缩[13];血卵泡刺激素(FSH)在婴儿期及儿童早期即已升高,6岁前逐渐降低,其后在正常青春期年龄又再次升高[14],FSH的升高与缺乏卵泡合成的抑制素负反馈抑制有关[14];血雌激素(E2)水平降低[13]。约1/3 TS患儿有自发青春期发育,多见于嵌合体核型者;其中少数可有月经来潮,但绝大多数最终发展为卵巢衰竭,自然怀孕率低(2%~5%)[15]。患儿常因生长迟缓、青春期无性征发育、原发性闭经等就诊。
除矮小和青春期发育异常外,TS患儿可有各种躯体畸形,见表2。约50%的TS患儿存在先天性心血管结构异常(如二叶式主动脉瓣、主动脉缩窄或扩张或主动脉壁夹层形成等)和非结构异常(如心电图电轴右偏,T波异常等)。其他系统异常,如肾脏畸形、骨骼畸形(表2)、糖耐量异常、甲状腺疾病等[15,16]。约29% TS患儿的甲状腺自身抗体[甲状腺过氧化酶抗体(TPOAb)、甲状腺球蛋白抗体(TgAb)]阳性,甲状腺自身抗体阳性的TS患儿甲状腺功能异常的例数远多于甲状腺自身抗体阴性的患儿[17]。
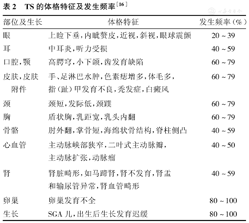
TS的体格特征及发生频率[16]
TS的体格特征及发生频率[16]
| 部位及生长 | 体格特征 | 发生频率(%) |
|---|---|---|
| 眼 | 上睑下垂,内眦赘皮,近视,斜视,眼球震颤 | 20~39 |
| 耳 | 中耳炎,听力受损 | 40~59 |
| 口腔,颚 | 高腭穹,小下颌,齿发育缺陷 | 60~79 |
| 皮肤,皮肤附件 | 手、足淋巴水肿,色素痣增多,体毛多,指(趾)甲发育不良,秃发症,白癜风 | 60~79 |
| 颈 | 颈短,发际低,颈蹼 | 60~79 |
| 胸 | 盾状胸,乳距宽,乳头内翻 | 60~79 |
| 骨骼 | 肘外翻,掌骨短,海绵状骨结构,脊柱侧凸 | 40~59 |
| 心血管 | 主动脉峡部狭窄,二叶式主动脉瓣,主动脉扩张,动脉瘤 | 40~50 |
| 肾 | 肾脏畸形,如马蹄肾,肾不发育,肾盂和输尿管异常,肾血管畸形 | 40~59 |
| 卵巢 | 卵巢发育不全 | 80~100 |
| 生长 | SGA儿,出生后生长发育迟缓 | 80~100 |
核型分析显示约5%的TS存在Y染色体物质,临床上患儿有男性化表现或存在染色体标志,应筛查Y染色体物质。存在Y染色体物质的患儿,有患性腺胚细胞瘤的风险,需切除性腺。
根据女性有特征性的体征和X染色体数目或结构异常[8](表1)可诊断为TS。但早期(如产前、新生儿期)诊断,及时检查可发现患儿可能存在的畸形,如主动脉缩窄及心、肾畸形等[7],并及时给予相应的处理。
30%~70%B超显示水囊状淋巴管瘤的胎儿,可能为TS。其他B超异常发现有颈背半透明、主动脉缩窄、短头(畸形)、肾脏畸形、羊水过多或过少和宫内生长障碍等[18]。母体血生物标记或DNA序列分析可用于发现TS胎儿:应用蛋白组学分析孕中期女性血标本,结果显示TS胎儿的母亲9种蛋白升高,即补体C1s(Complement C1s)、补体C3、聚集素(Clusterin)、重酒石酸苯丙胺(Afamin)、透明质酸结合蛋白2(Hyalouronan binding protein 2)、免疫球蛋白α-1链C区(Ig alpha-1 chain C region)、CD5抗原样蛋白(CD5 antigen-like protein)、性激素结合球蛋白(SHBG)和触珠蛋白(Haptoglobin);3种蛋白降低,即激肽原(Kininogen)、转甲状腺素蛋白(Transthyretin)和免疫球蛋白J链(Immunoglobulin J chain)[19],这些特异性的生物标记可能有助于产前诊断。最新报道应用大规模并行测序方法对孕期女性外周血进行分析,可明确怀孕早期胎儿是否患包括TS在内的所有常见非整倍性染色体异常的疾病[20]。
新生儿期早期诊断TS,可及时检查发现患儿可能存在的畸形,如主动脉缩窄及心、肾畸形等[7],并及时给予相应的处理。近10多年人们在探讨应用不同的分子方法(如PCR、PCR-限制性片段长度多态性等)早期筛查诊断TS。最新报道,用Real-time PCR方法检测血标本,计算芳香基硫酸酯酶基因与对照基因GAPDH的比值,该比值<0.7时诊断45,XO核型的特异性和敏感性均为100%[21]。该方法具有快速、敏感、特异等优点,可用于新生儿期筛查45,XO。不足的是基于PCR的方法尚不能可靠鉴定嵌合体或X染色体结构异常[18],而临床上40%的TS患者属此类异常。最新报道高通量焦磷酸测序方法用于TS诊断的敏感性和特异性分别为96%和97%[7],是有希望替代染色体核型分析的诊断工具。
淋巴水肿是婴儿期筛查TS的最常见的原因,而矮小则是儿童期筛查TS的常见原因[22],这些是诊断TS的重要临床线索。虽然部分TS患儿在婴儿和儿童期被诊断,然而大部分直到10岁后才被诊断[7],有报道约22%的TS患儿诊断被延迟至12岁以后[23]。因为TS的异常涉及人体的多系统和器官,所以,在明确TS诊断后需进行相关疾病的筛查,TS患儿诊断时的筛查项目见表3,并在其后长期的随访过程中,结合患儿的年龄发育阶段做相关项目的复查,复查项目见表4。


身高<第5百分位的TS患儿,4~5岁即可开始治疗。一项随机对照临床研究显示5~7年的GH治疗,可使TS的成年身高增长7.3 cm[24]。
建立于1987年的辉瑞国际成长数据库(Pfizer International Growth Database,KIGS),目前已收集来自全球50个不同国家共约5万例因各种生长障碍疾病而接受GH治疗的病例,对该数据库1146例生长接近成年身高的TS患儿的数据分析显示,该组患儿GH治疗前的生长速度为(4.6±1.9) cm/年,治疗第1年可增快至(7.6±1.9) cm/年;治疗前的身高标准差积分(SDS)为-3.4±1.0,治疗至接近成年身高时为-2.3±1.1,平均增加1.1 SDS[2]。
除单用GH外,人们还探讨各种联合治疗方案:(1)相对于当前一般在12岁才补充,有一项研究给TS患儿早至5岁(平均9.3岁)即开始补充小剂量乙炔雌二醇(EE2),结果单用GH组患儿身高增加了5 cm,而加用E2组身高多增加了2.1 cm[25],显示GH和E2的协同作用。(2)延迟青春期诱导至14~15岁,可使成年身高多增加约4 cm[15,26],但未在适当的年龄开始青春期诱导,对骨骼健康带来负面影响。(3)9岁以上的患儿或明显矮小者,可加用氧甲氢龙(Oxandrolone),剂量为0.05 mg/(kg·d),最大剂量2.50 mg/d,成年身高比单用GH的患儿平均多增长4 cm[26,27]。但需注意氧甲氢龙的肝功能异常、男性化等不良反应[22]。
疗效的影响因素:开始GH治疗时的身高较高及年龄较小,高父母身高,疗程较长以及GH剂量较高;一般在骨龄≥14岁或年身高增长<2 cm可考虑停止治疗;治疗期间应定期观察疗效,复查甲状腺功能、血生化,监测脊柱侧弯和脊柱后凸的发生[15]。
国内一般于TS患儿12岁后可开始E2替代治疗,模拟正常的性发育过程,促进乳房的发育和女性体征的形成。开始治疗时E2剂量应较小,如倍美力0.3125 mg/d(为成人剂量的1/6~1/4),用6~12个月;其后每3~6个月逐渐增加剂量至0.6250 mg/d[28]。根据治疗的反应及Tanner分期、骨龄、子宫的生长情况调整剂量,持续治疗1~2年[28]。第1次阴道出血发生后或E2治疗已经12~24个月考虑建立月经周期时,开始加用孕激素如安宫黄体酮,即每月第1-23天口服E2,于E2用药的第11天,开始加用安宫黄体酮5~10 mg/d,至第24天与E2同时停药。美国TS研究组的"TS临床指南"[8]的建议(表5),供国内参考。
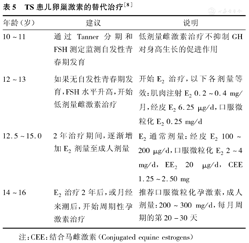
TS患儿卵巢激素的替代治疗[8]
TS患儿卵巢激素的替代治疗[8]
| 年龄(岁) | 建议 | 说明 |
|---|---|---|
| 10~11 | 通过Tanner分期和FSH测定监测自发性青春期发育 | 低剂量雌激素治疗不抑制GH对身高生长的促进作用 |
| 12~13 | 如果无自发性青春期发育,FSH水平升高,开始低剂量雌激素治疗 | 开始E2治疗,以下各剂量等效:肌肉注射E2 0.2~0.4 mg/月,经皮E2 6.25 μg/d,口服微粒化E2 0.25 mg/d |
| 12.5~15.0 | 2年治疗期间,逐渐增加E2剂量至成人剂量 | E2通常剂量:经皮E2 100~200 μg/d,口服微粒化E2 2~4 mg/d,EE2 20 μg/d,CEE 1.25~2.50 mg |
| 14~16 | E2治疗2年后,或月经来潮后,开始周期性孕激素治疗 | 推荐口服微粒化孕激素,成人剂量:200~300 mg/d,每月周期的第20-30天 |
注:CEE:结合马雌激素(Conjugated equine estrogens)
总之,TS是涉及遗传、发育、内分泌、心血管、心理和生殖等多学科的一个复杂的临床综合征。临床上尚需进一步完善由儿科内分泌、遗传、心血管、皮肤、发育、肾脏、眼科、整形外科、五官科和心理学等多专科医师组成的TS医疗团队,从而更好地为TS患儿服务。

























