
分析1例临床诊断为Walker-Warburg综合征(WWS)患儿的临床及遗传学特征。
收集该例患儿及父母的临床资料,提取患儿及其父母外周血基因组DNA,聚合酶链反应(PCR)扩增与此类疾病相关的5个候选基因包括POMT1、POMT2、POMGnT1、FKTN、FKRP基因的外显子,以琼脂糖凝胶电泳鉴定PCR产物,PCR产物纯化后DNA直接测序,确定基因突变的类型,分析基因型和表型的关系。
该患儿自幼智力运动发育落后,肌力、肌张力低下,早期出现关节挛缩,肌酸激酶中度升高,眼科检查疑诊青光眼,头颅核磁共振示侧脑室扩大,小脑、脑干发育不良,胼胝体发育不良,小脑囊肿,临床诊断为先天性肌营养不良伴眼脑畸形。进一步基因检测示患儿POMT1基因第5外显子发生了c.313C>T,p.Arg105Cys的错义突变,来自其父亲,第20外显子发生了c.2208delG,p.Trp736X的移码突变,来自其母亲。该患儿的2个突变均为已报道致病突变。
通过分子遗传学分析发现该患儿为POMT1基因的复合杂合突变,其突变基因分别来自其父母,符合WWS常染色体隐性遗传的特点,从基因水平可诊断为WWS,可为家庭提供遗传咨询和产前诊断。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
α-抗肌萎缩相关糖蛋白(α-DG)病是由于α-DG的糖基化缺陷导致的一组常染色体隐性遗传性先天性肌营养不良(CMD),包括福山型CMD(FCMD)、肌-眼-脑病(MEB)、Walker-Warburg综合征(WWS)、先天性肌营养不良1C型(MDC1C)和先天性肌营养不良1D型(MDC1D)[1]。WWS是α-DG病谱系中临床表型最严重的一种类型,除严重的肌无力及肌张力降低外尚伴严重的眼部受累和脑结构畸形。WWS在国内尚无报道,本研究对2009年就诊的1例WWS患儿的临床表型及候选基因进行分析,探讨该类疾病的基因型与临床表型的关系。
先证者,男,就诊时3个月。第2胎,第1产(第1胎自然流产),足月,孕期胎动少,因胎位不正、脐带绕颈剖宫产娩出,生后窒息(具体不详),哭声弱,肢体无力,3个月不会抬头,不会蹬被,可追光追物,逗笑差。就诊时体格检查:头围32.5 cm,肝肋下2.5 cm,脾未及,患儿消瘦,头左右活动差,四肢肌张力减低,肌力差,近端著,双膝关节、踝关节挛缩,双膝腱反射未引出,病理征(-)。眼科检查提示角膜大,疑诊青光眼。血清肌酸激酶(CK)2次检查结果分别为2 274.5 U/L和2 122.0 U/L,;心电图及超声心动图未见异常;头颅磁共振(MRI)示侧脑室扩大,小脑、脑干发育不良,胼胝体发育不良,小脑囊肿(图1)。本研究获患者家属的知情同意,并经医院伦理委员会讨论通过。
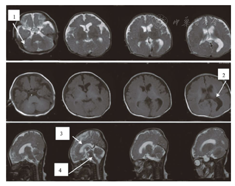
Brain magnetic resonance imaging of the patient with Walker-Warburg syndrome
注:1:小脑囊肿;2:侧脑室扩大;3:胼胝体发育不良;4:脑干、小脑发育不良 1:cerebellum cyst;2:extensive ventricles;3:corpus callosum dysplasia;4:dysplasia in the brainstem and cerebellum
Brain magnetic resonance imaging of the patient with Walker-Warburg syndrome
签署知情同意书后,采集患儿及其父母外周血5 mL,用Mill's蛋白盐析法提取基因组DNA。
聚合酶链反应(PCR)和PCR产物测序。从人类基因组数据库GenBank中获得POMT1、POMT2、POMGnT1、FKTN、FKRP的序列,应用加拿大Primer公司开发的Primer Premier 5.0软件,对编码区外显子及侧翼区域行PCR扩增引物设计。引物由北京天一辉远公司合成。反应体系20 μL中含2×PCR缓冲液,2.5 mmol/L dNTPs,10 pmol/L引物,1U Taq DNA聚合酶,50 ng模板DNA;反应条件:95 ℃热变性5 min后,95 ℃ 30 s,58 ℃ 20 s,72 ℃ 50 s,循环35次,接72 ℃延长5 min(不同基因反应条件略有不同)。20 g/L琼脂糖凝胶电泳鉴定PCR产物,经PCR纯化试剂盒(美国Qiagen公司)纯化,以PCR正向引物作为测序引物进行测序(北京天一辉远公司提供测序结果)。测序结果与人类基因组POMT1、POMT2、POMGnT1、FKTN、FKRP序列进行比较。使用DNASTAR软件分析结果。对测序结果异常的片段重新进行PCR扩增和反向测序,以验证结果的可靠性。
随访患儿智力运动发育严重落后,肌无力加重,喂养困难,于10个月时死亡,具体死因不详。
检测到患儿的POMT1基因第5外显子发生c.313C>T,p.Arg105Cys的杂合错义突变,来自其父亲;第20外显子发生移码突变,即c.2208delG,p.Trp736X,来自母亲,均为已报道致病突变,见图2。
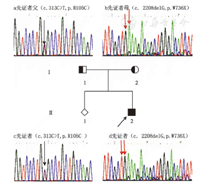
Genogram of proband and the result of POMT1 gene mutation test of Walker-Warburg syndrome
注:Ⅱ2:先证者;Ⅰ1:先证者父;Ⅰ2:先证者母;Ⅱ1:自然流产胎儿;a:先证者父POMT1的第5外显子错义突变c.313C>T,p.R105C;如箭头↓所示;b:先证者母POMT1的第20外显子移码突变c.2208delG,p.W736X,如箭头↓↓所示;c:先证者POMT1的第5外显子错义突变c.313C>T,p.R105C,如箭头↓所示;d:先证者POMT1的第20外显子移码突变c.2208delG,p.W736X,如箭头↓↓所示 Ⅱ2:proband;Ⅰ1:father of proband;Ⅰ2:mother of proband;Ⅱ1:spontaneous abortion fetal;a:father of proband has a missense mutation c.313C>T,p.R105C,just as the arrow showed;b:mother of proband has a frameshift mutation c.2208delG,p.W736X,just as the arrow showed;c:POMT1 gene of proband has a missense mutation c.313C>T,p.R105C,just as the arrow showed;d:POMT1 gene of proband has a frameshift mutation c.2208delG,p.W736X,just as the arrow showed
Genogram of proband and the result of POMT1 gene mutation test of Walker-Warburg syndrome
WWS是由Walker[2]于1942年首次报道,主要临床表现为出生时或出生后不久即起病,肌力、肌张力低下,运动发育落后伴智力发育迟滞,部分患儿伴癫 发作。WWS患儿常有较严重的眼部异常,如白内障,青光眼,高度近视,小眼畸形,水牛眼,视网膜脱离或发育不良,视神经萎缩或发育不良。脑部畸形包括严重的脑积水,小脑发育不良,脑干扁平,胼胝体发育不良,枕部脑膨出,脑白质显示髓鞘形成不良等,WWS病死率高,患儿多于3岁内死亡[3]。WWS患儿亦可发生唇裂和上腭裂[4]。
发作。WWS患儿常有较严重的眼部异常,如白内障,青光眼,高度近视,小眼畸形,水牛眼,视网膜脱离或发育不良,视神经萎缩或发育不良。脑部畸形包括严重的脑积水,小脑发育不良,脑干扁平,胼胝体发育不良,枕部脑膨出,脑白质显示髓鞘形成不良等,WWS病死率高,患儿多于3岁内死亡[3]。WWS患儿亦可发生唇裂和上腭裂[4]。
本例患儿孕期即有胎动少,出生后哭声弱,肢体无力,近端著,运动发育落后,3个月双膝关节及踝关节已有挛缩,腱反射减弱,查血清CK 2次均示明显升高,心电图和超声心动图正常,可考虑患有CMD。结合眼科检查疑诊青光眼,头颅MRI示侧脑室扩大,小脑、脑干发育不良,胼胝体发育不良,小脑囊肿,诊断符合CMD伴眼脑病变。结合患儿临床表现,作者对POMT1、POMT2、POMGNT1、FKTN及FKRP基因进行了分析,经测序证实在POMT1基因存在2个致病突变。随访患儿于10个月时死亡,进一步证实其患有CMD中较严重的类型,即WWS。
α-DG是肌营养不良糖蛋白聚糖复合物(DGC)的主要组成部分,是一个高度糖基化的细胞外环膜蛋白,在全身组织中广泛表达,在睾丸组织及胎儿的脑组织中表达尤其多。α-DG的糖基化主要发生在其黏蛋白样区域,糖基化的发生对于其与细胞外基质如肌肉组织中的Lamininα2、集聚蛋白及脑组织中的轴突蛋白相连十分重要[4,5,6,7]。O-甘露糖糖基化在α-抗肌萎缩相关糖蛋白糖基化中占有重要地位。POMT1编码的甘露糖转移酶1(POMT1)与POMT2编码的POMT2一起参与哺乳动物O-甘露糖基生物合成的第一步[8]。POMT1位于9q34.1,共有20个外显子,编码含747个氨基酸的POMT1共15个结构域,包括3个甘露糖转移酶(MIR)和12个跨膜区(TM),酶活性的丧失会导致α-DG的O-连接糖基化的缺陷。而α-DG糖基化的缺陷又会导致α-DG与其他的细胞外基质,如laminin-α2、laminin-β2及adhalin不能很好的连接,从而导致疾病的发生[1,9]。
患儿的突变分别来自其父母,其中c.313C>T,p.Arg105Cys来自父亲,该位点位于POMT1蛋白甘露糖转移酶的高度保守结构域PMT区域,已有文献报道此突变为致病突变,且相关表型也为典型的WWS,表现为头颅MRI示脑积水、胼胝体发育不良,小脑发育不良、脑白质异常信号,眼部异常,腓肠肌肥大等[10]。另一个突变为c.2208delG,p.Trp736X,位于跨膜区,位于POMT1酶活性结构域以外,来自患儿母亲,直接变为终止密码子,导致其后12个氨基酸未能表达,此突变亦有文献报道。文献中所报道为c.2208delG纯和突变,该患儿临床表型较轻,虽然出生后即发现肌无力和CK增高,但在22个月时已能扶坐,出生后42 d时查头颅MRI示后颅窝及中线结构正常,脑组织信号正常[11]。因此,本研究报道的患儿严重的临床表型与其位于酶活性结构域的p.Arg105Cys突变相关。患儿为POMT1基因复合杂合突变,临床表型符合WWS,随着对WWS的进一步研究,目前已报道导致WWS的致病基因除POMT1、POMT2外,FKTN、LARGE、ISPD、GTDC2、B3GNT1、B3GALNT2等基因突变也可导致此表型[12,13,14,15,16],但大样本的调查显示POMT1突变仍是导致WWS最常见的基因[12,17],本研究与文献报道相符。当然,近年文献报道,POMT1基因突变除导致WWS外,尚可能导致临床表型较轻的先天性肌营养不良伴智力障碍(CMD-MR),及肢带型肌营养不良伴智力障碍(LGMD-MR)[10,11],而同一种基因的突变导致不同的临床表型的原因可能是突变的位点不同,位于诸如POMT1酶活性结构域等高度保守区的突变其临床表型往往较重[10]。
综上所述,本研究报道的病例具有典型WWS的临床特点,结合遗传学分析,首次确诊了我国的WWS临床患者并明确了其临床特点。WWS病死率高,该患儿10个月时死亡,而患儿母亲前一次不明原因的自然流产很可能也是胎儿存在同样基因突变所致,因此,明确基因诊断可为患者家庭提供遗传咨询,避免同样孩子的出生。目前WWS尚无很好的治疗方法,良好的喂养及护理可适当延长患儿生命,康复治疗能够延缓患儿病情的进展,但这一疾病的生存时间仍较短,很多患儿在围生期就会死亡,其主要致死原因是肺部感染后呼吸和心脏衰竭,因此需加强护理,尽量减少肺部感染的发生。

























