
遗传性运动感觉性神经病是最常见的遗传性周围神经病,该病的特点为缓慢进展,肌无力及肌萎缩累及肢体近端及严重的足畸形可造成患者功能障碍。因为大多数患者在儿童期起病,所以,功能障碍的动态评估及规范化的治疗对改善患儿预后具有重要意义。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
遗传性运动感觉性神经病(hereditary motor and sensory neuropathy,HMSN)是由Charcot、Marie、Tooth 3人首次于1886年报导,至今已有120余年历史,因此,该病最初以3位科学家的名字命名为Charcot-Marie-Tooth disease(CMT,夏科-马里-图斯病),因患者多有腓骨肌萎缩,故又称腓骨肌萎缩症。目前CMT这一命名的应用较HMSN广泛。CMT是最常见的遗传性周围神经病,最新的国外流行病学调查显示其发病率约为1.2/10 000[1]。该病有2个发病高峰,为婴幼儿期及少年期,表现为多发的运动及感觉神经受累。该病早期表现为肢体远端肌肉的萎缩、无力及感觉减退,随着病情进展渐累及肢体近端。肌肉萎缩从足开始,表现为弓形足、锤状趾,随后渐累及小腿和大腿的下1/3,出现双下肢典型的"鹤腿征"。此阶段双上肢也常有不同程度受累。感觉神经的病变发展过程同上类似,主要累及双手和双足,并可伴感觉性共济失调。腱反射呈长度依赖性由远及近发展,逐渐减弱至消失。骨骼畸形主要累及双足,可有骨盆发育畸形及脊柱侧弯,发生率为25%[2,3]。
CMT1A是最常见的CMT类型,该型主要由周围神经髓鞘蛋白22(peripheral myelin protein 22,PMP22)基因重复突变所致,现已有数个相关家系被报道及随访。CMT1A临床表型常较典型,并较其他类型的CMT预后好,大多数患者终身保持行走能力。然而,CMT1A临床表型变化很大,少数患者表现为运动发育延迟及严重的骨骼畸形(包括脊柱侧弯),最终可能发展为严重近端肌肉无力,需助行器或轮椅助行。此外同一家系中的不同成员病情不一,即使同卵双胞胎病情也不一样[3,4,5]。
对于CMT其他类型的自然病程及预后目前研究不多。髓鞘蛋白0(myelin protein zero,MPZ)基因突变常导致2种相差很大的CMT。该基因突变所致的早发型脱髓鞘型的CMT1B或CMT3/Déjèrine-Sottas神经病,患者病情多较严重,部分患者最终可能需要轮椅。然而MPZ基因突变所致的晚发型轴索型CMT2,患者常起病晚,病情相对轻[6,7]。
线粒体融合蛋白2(mitofusion2,MFN2)基因突变所致的CMT2,可根据临床表型分为早发型(年龄<10岁)和晚发型(年龄≥10岁)。早发型患者病情通常较重,而晚发型患者大多病情较轻,病情进展前者较后者快。此外,在同一家系中同胞间功能障碍的严重程度相差不大,提示疾病的严重程度取决于MFN2基因突变位点[8]。
CMTX1为X连锁遗传,常由间隙连接蛋白β1(gap junction β1,GJB1)基因突变所致。该型在男性中常于10~20岁起病,病情进展隐匿,常在晚期导致严重的残疾。CMTX1的杂合子女性可无任何症状,仅有轻度的临床及神经电生理异常提示CMT。但在女性CMTX1中也有极少数病例病情很严重,这种情况与X染色体失活偏移有关[9]。
常染色隐性遗传的CMT(轴索型AR-CMT2及脱髓鞘型CMT4)病情常较显性遗传者严重。该型多起病早,功能障碍更严重[10]。其中神经节苷脂诱导分化相关蛋白1(ganglioside-induced differentiation-associated protein 1,GDAP1)基因突变所致的AR-CMT患者常伴手运动功能丧失,患者严重的近端肌无力最终常使其在20~30岁时依赖轮椅[11]。CMT患者如同时携带2种致病基因病情常更严重[12]。
关于CMT患者功能障碍的评估包括疼痛评估、肺功能评估、生活质量评估及功能障碍程度的评估等。CMT患者功能残障程度的评估目前研究较多,应用前景较好。在2005年第1版CMT神经病评分量表出现前,有关的功能障碍评估方法除可用于CMT外,还可用于其他慢性周围神经病患者甚或其他慢性病患者,并各有其侧重面。如功能残疾度(function disability score,FDS)及Rankin评分主要从行走能力评估CMT患者的功能残疾度,其中Rankin评分主要应用于脑卒中患者双下肢能力的评估。运动功能残疾度(motor neurological disability score,MNDS)主要是从四肢肌肉萎缩及肌无力的角度进行评估。神经功能障碍评分(neurological impairment score,NIS)及全面性神经障碍评分(global neurological disability score,GNDS)所涉及的项目较相近,通过全面的神经系统检查从肌萎缩、肌力、感觉、腱反射方面对患者进行功能障碍评估[5,13]。足姿势指数(foot posture index,FPI)主要用于评估患者足畸形程度[14]。
足畸形是神经肌肉病的常见的表现,且足畸形的程度直接影响到患者功能障碍的程度。目前对足畸形病因的研究发现,进行性发展的足部畸形主要是由CMT所致(52.8%)[15]。患者正常的行走、跑跳能力需正常的足部的解剖结构及肌力。Rose等[16]对CMT患儿足部肌力及足畸形的研究发现,CMT患儿同健康同龄儿足比较,其足畸形常在5岁以后渐明显,而足部肌力减退则在2~4岁时就出现,因此,早期患儿足畸形表现为足下垂。
目前可用于CMT患儿足畸形的评估方法主要有Coleman木板试验或斜砖试验及负重弓步试验等。Coleman木板试验或斜砖试验主要用于评估患者后足内翻及前足的内旋程度,如能纠正说明患者后足尚有一定的柔韧性,后足内翻畸形可通过矫形器或足部软组织手术纠正,如不能纠正则只能通过骨性手术纠正。负重弓步试验主要用于评估患儿足背屈程度[16,17]。
近年来骨科医师常用FPI评估CMT患儿足畸形,FPI分为-12~12分,-12~-1分提示高足弓畸形,0~5分提示足姿势正常,6~12分提示扁平足。Burns等[18]对CMT1A患儿足畸形的动态观察中发现,CMT患儿的足畸形随着年龄增大发生率增高,FPI与患儿身高、体质量、年龄呈负相关。负重位X线检查评估患儿前足、中足、后足的畸形,也是近年来逐渐兴起的一种评估足畸形的手段。相关的研究发现,CMT患儿前足、中足、后足水平所测得的各个解剖角度与健康同龄儿比较差异均有统计学意义,CMT患儿足畸形的程度与患儿FDS及CMT神经病评分量表(CMTNS)直接相关[14]。Ferrarin等[19]对CMT1A患儿进行18个月的步态随访研究发现,通过使用步态分析仪评估患儿的行走能力及下肢的功能方面较CMTNS此类的评估量表敏感,且可用于评估足部畸形矫正手术的效果。
国际上关于CMT患者功能障碍评估的量表在2005年开始应用CMTNS,见表1。该量表在成人及10岁以上儿童中可行性及敏感性均较好。该量表与其他项目包括行走指数、自我评估、手功能、9孔插棒试验及神经病损害评分(Neuropathy Impairment score,NIS)在评估残疾程度方面具有较好的一致性。CMTNS根据患者所得分情况分为轻度(≤10分),中度(11~20分),重度(≥21分)。该量表诞生后即用于CMT病情变化的研究[20]。但Haberlova和Seeman[21]对20例年龄较小的CMT1A儿童应用CMTNS评分发现,所有患者的CMTNS均低于10分,均为轻度,患者的得分主要是来自神经电生理异常。同时Pagliano等[13]研究也提示对于年龄较小的CMT患者应用CMTNS评估病情敏感度有限,对于年龄较小的CMT患者可能需更科学的方法来评估病情。
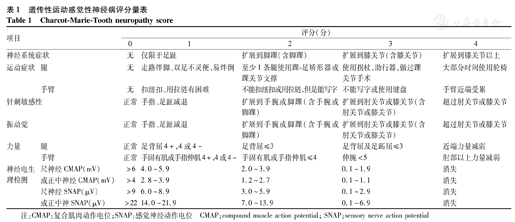
遗传性运动感觉性神经病评分量表
Charcot-Marie-Tooth neuropathy score
遗传性运动感觉性神经病评分量表
Charcot-Marie-Tooth neuropathy score
| 项目 | 评分(分) | |||||
|---|---|---|---|---|---|---|
| 神经系统症状 | 0 | 1 | 2 | 3 | 4 | |
| 运动症状 | 腿 | 无 | 走路绊脚、双足不灵便,易绊倒 | 至少1条腿使用踝-足矫形器或踝关节支撑 | 使用拐杖,助行器,做过踝关节手术 | 大部分时间使用轮椅 |
| 手臂 | 无 | 扣纽扣、用拉链有困难 | 不能扣纽扣或用拉链,但是能写字 | 不能写字或使用键盘 | 手臂近端受累 | |
| 针刺敏感性 | 正常 | 手指、足趾减退 | 扩展到手腕或脚踝(含手腕或脚踝) | 扩展到肘关节或膝关节(含肘关节或膝关节) | 超过肘关节或膝关节 | |
| 振动觉 | 正常 | 手指、足趾减退 | 扩展到手腕或脚踝(含手腕或脚踝) | 扩展到肘关节或膝关节(含肘关节或膝关节) | 超过肘关节或膝关节 | |
| 力量 | 腿 | 正常 | 足背屈4+,4或4- | 足背屈≤3 | 足背屈及足跖屈≤3 | 近端力量减弱 |
| 手臂 | 正常 | 手固有肌或手指伸肌4+,4或4- | 手固有肌或手指伸肌≤4 | 伸腕<5 | 肘部以上力量减弱 | |
| 神经电生理检测 | 尺神经CMAP(mV) | >6 | 4.0~5.9 | 2.0~3.9 | 0.1~1.9 | 消失 |
| 或正中神经CMAP(mV) | >4 | 2.8~3.9 | 1.2~2.7 | 0.1~1.1 | 消失 | |
| 尺神经SNAP(μV) | >9 | 6.0~8.9 | 3.0~5.9 | 0.1~2.9 | 消失 | |
| 或正中神SNAP(μV) | >22 | 14.0~21.9 | 7.0~13.9 | 0.1~6.9 | 消失 | |
注:CMAP:复合肌肉动作电位;SNAP:感觉神经动作电位 CMAP:compound muscle action potential; SNAP:sensory nerve action potential
尽管CMTNS对评估CMT严重程度效果较好,但近些年在使用该量表时发现量表中的某些项目存在地板效应(ceiling or floor effect)。第168届欧洲神经肌肉病中心(ENMC)国际研讨会提出对CMTNS进行调整,并将评估方法标准化,从而提高评估量表的敏感性[22]。2011年CMTNSv2诞生,该量表避免了CMTNS的地板效应,但2个量表在评估的敏感性方面无明显差异[23]。
鉴于CMTNS在年龄较小的CMT患者中敏感性可能有限,2012年出现了CMT儿科量表,该表适用于3~18岁确诊CMT的患儿。包括体力、灵活度、步态、平衡、力量、耐力的评估,共11个评估项目,总分44分。该量表适用于各型CMT[24]。2013年最新的一项研究表明,在儿童功能障碍的评估中,CMT儿童量表与CMTNSv2的评估结果较一致[25]。
CMT为一慢性进行性发展的神经肌肉病,可导致患儿功能障碍,包括行走、跑跳能力等,严重影响其生活质量,规范化的疾病管理对CMT患儿长期预后的改善具有重大意义。功能障碍评估方面CMT儿童量表已推出,CMTNS已较广泛地应用于临床及相关的科研工作中,但这2种量表均有一定的局限性,希望今后能开发出一款既适合儿童也适用于成年人的量表。

























