
探讨结节性硬化症(TSC)TSC1/TSC2突变基因型与表型的相关性。
应用高通量捕获二代测序技术检测19例中国儿童TSC基因突变类型,并对其基因型(包括新发突变)与临床表型进行分析。
在19例TSC患儿中,4例为TSC1基因突变,13例为TSC2基因突变,2例突变基因不明;其中TSC2/TSC1基因突变率之比为3.4/1,共发现6种新突变基因位点。TSC1基因突变组有癫 发作3例(75%,3/4例),TSC2基因突变组有癫
发作3例(75%,3/4例),TSC2基因突变组有癫 发作10例(77%,10/13例);TSC1基因突变组癫
发作10例(77%,10/13例);TSC1基因突变组癫 发作难以控制者1例(25%,1/4例),TSC2基因突变组癫
发作难以控制者1例(25%,1/4例),TSC2基因突变组癫 发作难以控制者4例(31%,4/13例),TSC1和TSC2基因突变组的癫
发作难以控制者4例(31%,4/13例),TSC1和TSC2基因突变组的癫 发作及严重程度差异均无统计学意义(P=0.480 7、0.462 2)。85%(11/13例)TSC2基因突变组患儿有智力障碍,高于TSC1基因突变组(50%,2/4例);TSC2基因突变组有中重度智力障碍者7例(54%,7/13例),高于TSC1基因突变组(25%,1/4例)。TSC2突变基因型出现皮肤、心脏和肾脏损害的临床表型可能多于TSC1突变基因型。
发作及严重程度差异均无统计学意义(P=0.480 7、0.462 2)。85%(11/13例)TSC2基因突变组患儿有智力障碍,高于TSC1基因突变组(50%,2/4例);TSC2基因突变组有中重度智力障碍者7例(54%,7/13例),高于TSC1基因突变组(25%,1/4例)。TSC2突变基因型出现皮肤、心脏和肾脏损害的临床表型可能多于TSC1突变基因型。
TSC2基因突变可能是TSC患儿主要的分子发病机制,TSC2基因发生突变的患儿其临床表型更严重,较易发生癫 、智力障碍、面部血管纤维瘤及肾错构瘤等,但尚需进一步进行多中心、大样本研究证实。
、智力障碍、面部血管纤维瘤及肾错构瘤等,但尚需进一步进行多中心、大样本研究证实。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
结节性硬化症(TSC)属于常染色体显性遗传性神经皮肤综合征,TSC1和TSC2是该病主要的致病基因,虽然TSC基因型和表型的关系在一些国家已有报道[1,2,3],但国内仅见关于TSC伴婴儿痉挛表型与基因型关系的分析[4],突变的多样性、表型特征和基因型的相关性仍不清楚。因此,本研究应用高通量捕获测序技术检测19例中国儿童TSC基因突变类型,并对其基因型(包括新发突变)与临床表型进行分析。
武汉市儿童医院神经内科2011年1月至2014年2月收治的TSC患儿19例。其中男13例,女6例,男女之比为2.2:1.0;起病年龄为50 d~1.5岁。具有TSC家族史者4例,散发病例15例,诊断标准符合美国TSC联盟制定的标准[1]。收集患儿详细的病史资料,包括发病年龄、家族史、皮肤等一般体格检查及神经系统检查资料。患儿均接受脑电图、头颅磁共振(MRI)检查,心脏、腹部、肾脏彩色多普勒超声检查,眼底Ret Ham检查及智力测试筛查,患儿均进行基因检测。患儿家属均签署了知情同意书,并经医院伦理委员会审批通过。基因检测是由武汉东西湖高新技术开发区华大基因公司完成的。
对中枢神经系统损害临床表型的严重程度进行分级[5]。(1)精神发育迟滞(MR):4岁以上采用韦氏智力量表(WISC),根据测试的智商(IQ)结果将智力水平分为0~3级。0级:正常和边缘,IQ≥70分;1级:轻度异常,IQ 50~69分;2级:中重度异常,IQ<50分。4岁以下采用Gesell发育量表,根据测试的发育商(DQ)结果将智力水平分级。0级:正常和边缘,DQ≥75分;1级:轻度异常,DQ 55~75分;2级:中重度异常,DQ<55分。(2)癫 (根据有无癫
(根据有无癫 发作和药物控制情况分级):0级:无癫
发作和药物控制情况分级):0级:无癫 发作;1级:治疗后发作控制良好;2级:治疗后控制不佳。(3)室管膜下结节,皮质或皮质下结节:根据头颅MRI显示的结节数目分为3级:0级=无神经影像学损害;1级<3个;2级≥3个。
发作;1级:治疗后发作控制良好;2级:治疗后控制不佳。(3)室管膜下结节,皮质或皮质下结节:根据头颅MRI显示的结节数目分为3级:0级=无神经影像学损害;1级<3个;2级≥3个。
抽取受试者及对照者静脉乙二胺四乙酸抗凝血5 mL,标准流程提取基因组DNA(QIAamp DNA Blood Midi Kit,Qiagen,Hilden,Germany)。利用Covafis S2超声波仪(Massachusetts,USA)将基因组DNA打断成200~250 bp的片段,随后进行Ampure Beads纯化,将1 μg提纯的DNA片段进行末端修复、加"A"以及加接头反应,从而完成单个患者的DNA建库。Non–Captured样品进行连接介导PCR(LM–PCR)反应,纯化,利用定制的基因片段捕获芯片(Roche NimbleGen,Madison,USA)与10~20个标记好的患者DNA库42 ℃杂交72 h,杂交结束后进行芯片的洗涤和洗脱反应,随后进行Captured样品的LM–PCR反应。该芯片共捕获TSC相关基因的全部外显子加上外显子两侧翼的10 bp。样品经Agilent 2100 Bioanalyzer(Agilent Technologies,Inc.,Santa Clara,CA,USA)和ABI StepOne(Applied Biosystems Life Technologies,Foster City,CA,USA)进行富集度的检测,最后利用高通量测序仪Illumina HiSeq2000 Analyzers(Illumina,SanDiego,USA)连续双向测序90个循环,用Illumina Pipeline software(version 1.3.4)读出原始测序数据。
数据下机后进入信息分析部分。首先对下机的原始数据(Raw reads)进行测序质量评估,去除低质量以及被接头污染的reads。随后用BWA软件(Burrows Wheeler Aligner)[7]与HG19进行序列比对,与此同时进行序列捕获效果评价,用SOAPsnp[8]软件和Samtools[9]软件分别进行SNV(single nucletide variant)和Indel(insertion and deletion)的查询,生成目标区域碱基多态性结果,随后进行数据库(NCBI dbSNP,HapMap,1 000 human genome dataset和database of 100 Chinese healthy adults)的比对,并对找出的可疑突变进行注释、筛选。
对于所有发现的致病突变,在其所在片段上下游设计引物。进行PCR扩增,对产物做Sanger测序,所得结果与基因标准序列进行比对,从而验证基因芯片捕获和高通量测序的结果。
采用SAS 9.2软件进行统计学分析,运用Fisher's精确概率法对分类变量资料判别分析,比较TSC1基因突变和TSC2基因突变患儿临床表型之间的差异,P<0.05为差异有统计学意义。
如表1所示,19例TSC患儿中有13种TSC2基因突变及4种TSC1基因突变,TSC基因突变总检测率89.5%(17/19例),TSC2/TSC1基因突变率之比为3.4,2例未检测到TSC1或TSC2基因位点突变。
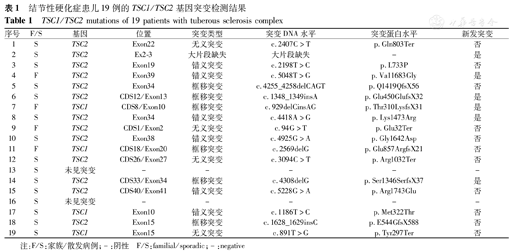
结节性硬化症患儿19例的TSC1/TSC2基因突变检测结果
TSC1/TSC2 mutations of 19 patients with tuberous sclerosis complex
结节性硬化症患儿19例的TSC1/TSC2基因突变检测结果
TSC1/TSC2 mutations of 19 patients with tuberous sclerosis complex
| 序号 | F/S | 基因 | 位置 | 突变类型 | 突变DNA水平 | 突变蛋白水平 | 新发突变 |
|---|---|---|---|---|---|---|---|
| 1 | S | TSC2 | Exon22 | 无义突变 | c.2407C>T | p.Gln803Ter | 否 |
| 2 | S | TSC2 | Ex2–3 | 大片段缺失 | 大片段缺失 | – | 是 |
| 3 | S | TSC2 | Exon19 | 错义突变 | c.2198T>C | p.L733P | 否 |
| 4 | F | TSC2 | Exon39 | 错义突变 | c.5048T>G | p.Va11683Gly | 是 |
| 5 | S | TSC2 | Exon34 | 框移突变 | c.4255_4258delCAGT | p.Q1419QfsX56 | 否 |
| 6 | S | TSC2 | CDS12/Exon13 | 框移突变 | c.1348_1349insA | p.Glu450GlufsX32 | 是 |
| 7 | F | TSC1 | CDS8/Exon10 | 框移突变 | c.929delCinsAG | p.Thr310LysfsX31 | 是 |
| 8 | S | TSC2 | Exon34 | 错义突变 | c.4418A>G | p.Lys1473Arg | 是 |
| 9 | F | TSC2 | CDS1/Exon2 | 无义突变 | c.94G>T | p.Glu32Ter | 否 |
| 10 | S | TSC2 | Exon38 | 错义突变 | c.4925G>A | p.Gly1642Asp | 否 |
| 11 | F | TSC1 | CDS18/Exon20 | 框移突变 | c.2569delG | p.Glu857ArgfsX21 | 否 |
| 12 | S | TSC2 | CDS26/Exon27 | 无义突变 | c.3094C>T | p.Arg1032Ter | 否 |
| 13 | S | 未见突变 | – | – | – | – | – |
| 14 | S | TSC2 | CDS33/Exon34 | 框移突变 | c.4308delG | p.Ser1346SerfsX37 | 是 |
| 15 | S | TSC2 | CDS40/Exon41 | 错义突变 | c.5228G>A | p.Arg1743Glu | 否 |
| 16 | S | 未见突变 | – | – | – | – | – |
| 17 | S | TSC1 | Exon10 | 错义突变 | c.1186T>C | p.Met322Thr | 否 |
| 18 | S | TSC2 | Exon15 | 框移突变 | c.1628_1629insC | p.E544GfsX588 | 否 |
| 19 | S | TSC1 | Exon15 | 无义突变 | c.891T>G | p.Tyr297Ter | 否 |
注:F/S:家族/散发病例;–:阴性 F/S:familial/sporadic;–:negative
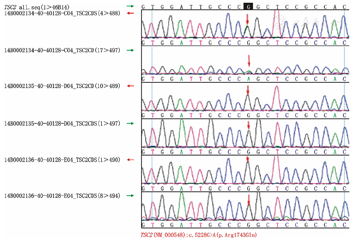
13种TSC2基因突变中,含5种新发突变,有TSC家族史者2例,余为散发病例。其中TSC2基因突变中错义或无义突变8种,框移突变4种,大片段缺失1种。1、6、9、14号患儿的TSC2基因Exon22、CDS12/Exon13、CDS1/Exon2、CDS33/Exon34 (c.2407C>T,c.1348_1349insA,c.94G>T,c.4038delG)均引起氨基酸序列提前终止编码。15号病例其TSC2基因的编码区(CDS40/Exon41)发现1个错义突变c.5228G>A(p.Arg1743Gln),为杂合子;其在人群中发生的频率较低,经SIFT和Polyphen对其进行蛋白功能预测,结果均显示为有害;有文献报道在患者中检出此突变位点,但未对其致病性做明确定论[10],进一步行Sanger测序峰图证实,其在TSC2基因编码区确实存在错义突变c.5228G>A(p.Arg1743Gln),为杂合子,与高通量检测结果一致,表明错义突变c.5228G>A应为可疑致病性突变(图1、图2)。4种TSC1基因突变中,仅见1种新发突变,其中TSC1基因突变中分别含错义和无义突变各1种,框移突变2种,未见大片段缺失。有TSC家族史者2例,散发病例2例。11、19号患儿的TSC1基因区CDS18/Exon20、Exon15发现1个框移突变c.2569delG(p.Glu857ArgfsX21)和1个无义突变c.891T>G造成TSC1氨基酸编码提前终止。
见表2、表3。17例TSC基因突变(TSC2突变13例,TSC1突变4例)的TSC患儿中,有癫 发作13例(76%,13/17例),其中TSC2基因突变组10例(77%,10/13例),TSC1基因突变组3例(75%,3/4例),TSC2基因突变组癫
发作13例(76%,13/17例),其中TSC2基因突变组10例(77%,10/13例),TSC1基因突变组3例(75%,3/4例),TSC2基因突变组癫 发作达2级(即药物治疗难以控制)4例(31%,4/13例),TSC1基因突变组1例(25%,1/4例),从癫
发作达2级(即药物治疗难以控制)4例(31%,4/13例),TSC1基因突变组1例(25%,1/4例),从癫 发生率和严重程度上比较,2组差异无统计学意义(P=0.480 7和0.462 2);首发癫
发生率和严重程度上比较,2组差异无统计学意义(P=0.480 7和0.462 2);首发癫 的年龄为50 d,发病年龄分布主要集中于1岁以内的患儿,共12例,1岁以上出现癫
的年龄为50 d,发病年龄分布主要集中于1岁以内的患儿,共12例,1岁以上出现癫 发作仅1例;发作形式表现为局限性发作者8例,其中1例发展为婴儿痉挛,1例呈局灶性癫
发作仅1例;发作形式表现为局限性发作者8例,其中1例发展为婴儿痉挛,1例呈局灶性癫 持续状态,早期即以婴儿痉挛起病者4例(21%,4/19例),所有诊断婴儿痉挛的患儿TSC突变基因型均为TSC2突变。
持续状态,早期即以婴儿痉挛起病者4例(21%,4/19例),所有诊断婴儿痉挛的患儿TSC突变基因型均为TSC2突变。
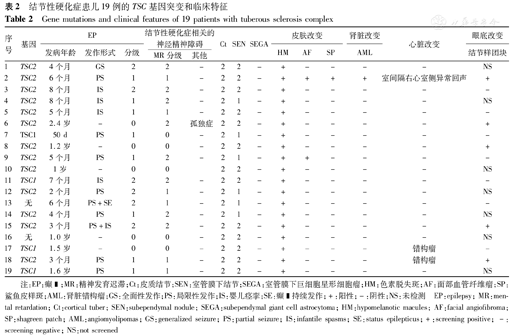
结节性硬化症患儿19例的TSC基因突变和临床特征
Gene mutations and clinical features of 19 patients with tuberous sclerosis complex
结节性硬化症患儿19例的TSC基因突变和临床特征
Gene mutations and clinical features of 19 patients with tuberous sclerosis complex
| 序号 | 基因 | EP | 结节性硬化症相关的神经精神障碍 | Ct | SEN | SEGA | 皮肤改变 | 肾脏改变 | 心脏改变 | 眼底改变 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 发病年龄 | 发作形式 | 分级 | MR分级 | 其他 | HM | AF | SP | AML | 结节样团块 | ||||||
| 1 | TSC2 | 4个月 | GS | 2 | 2 | – | 2 | 2 | – | + | – | – | – | – | NS |
| 2 | TSC2 | 6个月 | PS | 1 | 1 | – | 2 | 2 | – | + | + | + | + | 室间隔右心室侧异常回声 | + |
| 3 | TSC2 | 8个月 | IS | 2 | 2 | – | 2 | 2 | – | + | – | – | – | – | – |
| 4 | TSC2 | 8个月 | IS | 1 | 2 | – | 2 | 1 | – | + | – | – | – | – | NS |
| 5 | TSC2 | 5个月 | IS | 1 | 1 | – | 2 | 2 | – | + | – | – | – | – | – |
| 6 | TSC2 | 2.4岁 | – | 0 | 2 | 孤独症 | 2 | 2 | – | + | – | – | – | – | + |
| 7 | TSC1 | 50 d | PS | 1 | 0 | – | 2 | 1 | – | + | – | – | – | – | – |
| 8 | TSC2 | 1.2岁 | – | 0 | 0 | – | 2 | 2 | – | + | – | – | – | – | + |
| 9 | TSC2 | 5个月 | PS | 1 | 2 | – | 2 | 1 | – | + | + | – | – | – | – |
| 10 | TSC2 | 1岁 | – | 0 | 0 | 2 | 2 | – | + | – | – | – | – | NS | |
| 11 | TSC1 | 7个月 | IS | 2 | 2 | – | 2 | 2 | – | + | – | – | – | – | – |
| 12 | TSC2 | 2个月 | PS | 2 | 1 | – | 2 | 1 | – | + | – | – | – | – | NS |
| 13 | 无 | 6个月 | PS+SE | 2 | 1 | – | 2 | 1 | – | + | – | – | – | – | – |
| 14 | TSC2 | 4个月 | PS | 1 | 2 | – | 2 | 1 | – | + | – | – | – | – | NS |
| 15 | TSC2 | 3个月 | PS+IS | 2 | 2 | – | 2 | 2 | – | + | – | – | – | – | + |
| 16 | 无 | 1.0岁 | – | 0 | 0 | – | 2 | 2 | – | + | – | – | – | – | NS |
| 17 | TSC1 | 1.5岁 | – | 0 | 0 | – | 2 | 2 | – | + | – | – | – | 错构瘤 | – |
| 18 | TSC2 | 3个月 | PS | 1 | 1 | – | 2 | 2 | – | + | – | – | – | 错构瘤 | + |
| 19 | TSC1 | 1.6岁 | PS | 1 | 1 | – | 2 | 2 | – | + | – | – | – | – | NS |
注:EP:癫 ;MR:精神发育迟滞;Ct:皮质结节;SEN:室管膜下结节;SEGA:室管膜下巨细胞星形细胞瘤;HM:色素脱失斑;AF:面部血管纤维瘤;SP:鲨鱼皮样斑;AML:肾脏错构瘤;GS:全面性发作;PS:局限性发作;IS:婴儿痉挛;SE:癫
;MR:精神发育迟滞;Ct:皮质结节;SEN:室管膜下结节;SEGA:室管膜下巨细胞星形细胞瘤;HM:色素脱失斑;AF:面部血管纤维瘤;SP:鲨鱼皮样斑;AML:肾脏错构瘤;GS:全面性发作;PS:局限性发作;IS:婴儿痉挛;SE:癫 持续发作;+:阳性;–:阴性;NS:未检测 EP:epilepsy; MR:mental retardation; Ct:cortical tuber; SEN:subependymal nodule; SEGA:subependymal giant cell astrocytoma; HM:hypomelanotic macules; AF:facial angiofibroma; SP:shagreen patch; AML:angiomyolipomas; GS:generalized seizure; PS:partial seizure; IS:infantile spasms; SE:status epilepticus;+:screening positive;–:screening negative; NS:not screened
持续发作;+:阳性;–:阴性;NS:未检测 EP:epilepsy; MR:mental retardation; Ct:cortical tuber; SEN:subependymal nodule; SEGA:subependymal giant cell astrocytoma; HM:hypomelanotic macules; AF:facial angiofibroma; SP:shagreen patch; AML:angiomyolipomas; GS:generalized seizure; PS:partial seizure; IS:infantile spasms; SE:status epilepticus;+:screening positive;–:screening negative; NS:not screened
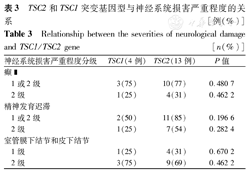
TSC2和TSC1突变基因型与神经系统损害严重程度的关系[例(%)]
Relationship between the severities of neurological damage and TSC1/TSC2 gene[n(%)]
TSC2和TSC1突变基因型与神经系统损害严重程度的关系[例(%)]
Relationship between the severities of neurological damage and TSC1/TSC2 gene[n(%)]
| 神经系统损害严重程度分级 | TSC1(4例) | TSC2(13例) | P值 | |
|---|---|---|---|---|
癫 | ||||
| 1或2级 | 3(75) | 10(77) | 0.480 7 | |
| 2级 | 1(25) | 4(31) | 0.462 2 | |
| 精神发育迟滞 | ||||
| 1或2级 | 2(50) | 11(85) | 0.196 6 | |
| 2级 | 1(25) | 7(54) | 0.282 4 | |
| 室管膜下结节和皮下结节 | ||||
| 1级 | 1(25) | 4(31) | 0.670 2 | |
| 2级 | 3(75) | 9(69) | 0.462 2 | |
17例TSC基因突变患儿中MR者13例(76%),TSC2基因突变组MR发生率85%(11/13例)高于TSC1基因突变组(50%,2/4例),TSC2基因突变组MR达2级者有7例(54%,7/13例)高于TSC1基因突变组的1例(25%,1/4例),但2组MR发生率和严重程度上相比差异无统计学意义(P=0.196 6,0.282 4);13例有癫 发作的患儿中,12例(92%)出现智力障碍,4例无癫
发作的患儿中,12例(92%)出现智力障碍,4例无癫 发作的TSC患儿中,仅1例TSC2基因突变患儿因诊断孤独症表现为智能发育迟缓,余3例均无智力障碍(75%,3/4例)。
发作的TSC患儿中,仅1例TSC2基因突变患儿因诊断孤独症表现为智能发育迟缓,余3例均无智力障碍(75%,3/4例)。
19例TSC患儿中,头颅MRI均可见皮质或皮质下数目≥3个的多发结节异常信号影,未见到室管膜下巨细胞星形细胞瘤,室管膜下结节状短T1、T2信号影仅有6例<3个,从神经影像学结节数目分级严重程度比较,TSC2基因突变组和TSC1基因突变组无统计学意义(P>0.05)。
19例TSC患儿中,皮肤均可见≥3块的色素脱失斑,均未见肾囊肿,因年龄小均未行肺部相关检查。2、9号TSC2突变患儿随访第3年时面部出现血管纤维瘤,其中2号背部皮肤伴有鲨鱼皮斑,心脏彩超为室间隔右心室侧异常回声,性质不详,双肾彩超显示存在错构瘤;2例(17、18号)胎儿彩超显示心脏横纹肌瘤,基因型分别是TSC1和TSC2突变;19例中12例行眼底Ret Ham检测,其中5例(42%,5/12例)基因型均为TSC2突变患儿,眼底可见大小不等的白色团块或白斑。
TSC是一种以多器官的组织缺陷和错构瘤为特征的常染色体显性遗传性神经皮肤综合征,TSC1和TSC2 2个肿瘤抑制基因是该病的致病基因。目前已发现664种TSC1基因突变及1 807种TSC2基因突变。对TSC患者进行基因分析,可发现80%~85%的患者存在TSC1或TSC2基因突变,仅15%~20%临床确诊TSC的患者,检测不到TSC基因突变,虽然散发病例中TSC2基因突变发生率明显高于TSC1,可能与肿瘤抑制基因相邻区域的杂合子缺失(LOH)较少发生在TSC1位点有关,但不同种族之间TSC2/TSC1基因突变比例有所不同,欧美人群TSC患者TSC2/TSC1基因突变比率为3.4~5.6[10,11,12],而Niida等[13]发现日本TSC患者人群中TSC1基因突变率高于欧美国家,其TSC2/TSC1基因突变比率仅为1.9,其原因不明,推测与TSC1基因突变的患者临床表型较不严重而未到医院就诊有关。本研究对19例TSC患儿基因型进行检测,发现有13种TSC2基因突变及4种TSC1基因突变,TSC基因突变总检测率89.5%,TSC2/TSC1基因突变率之比为3.4,类似于欧美人群[14],并发现6种新发突变。2例未检测出TSC基因突变患儿分别在TSC1和TSC2基因编码区发现1个剪切突变(c.2626–4_–3insTT,c.5161–9C>T)的多态性位点。对于临床已确诊,而基因检测不到TSC突变的原因可能是致病性突变位点隐藏在3`UTR序列中,而目前3'UTR序列功能尚不明确。另外,仅TSC1或TSC2的致病性突变方能作为基因诊断标准,但本研究有1例患儿(15号)通过高通量检测技术发现其TSC2基因的编码区(CDS40/Exon41)1个错义突变c.5228G>A(p.Arg1743Gln),为杂合子,并经进一步行Sanger测序峰图证实,虽仅有1篇文献报告在患者中检出此突变位点,并未对其致病性做明确定论,但经SIFT和Polyphen对其进行蛋白功能预测,结果均显示为有害,仍考虑其为可疑致病性突变,有待后续样本进一步证实。
神经系统损害包括癫 、智力障碍和孤独症均是最常被关注的TSC病变。有文献显示85%的TSC患者会出现癫
、智力障碍和孤独症均是最常被关注的TSC病变。有文献显示85%的TSC患者会出现癫 发作,其中婴儿痉挛占38%,63%的癫
发作,其中婴儿痉挛占38%,63%的癫 发作发生在出生1年以内,发作类型多样[15]。本研究确定TSC基因突变的17例TSC患儿中,出现癫
发作发生在出生1年以内,发作类型多样[15]。本研究确定TSC基因突变的17例TSC患儿中,出现癫 发作的占76%,71%TSC患儿发病年龄分布主要集中于1岁以内,发作形式主要表现为局限性发作,早期即以婴儿痉挛起病的占21%,其中1例由局限性发作进展而来。值得注意的是,本研究中4例婴儿痉挛的患儿TSC突变基因型均为TSC2突变,推测伴有婴儿痉挛的TSC患儿可能与TSC2突变基因型相关。本研究比较了TSC1和TSC2基因突变2组患儿的癫
发作的占76%,71%TSC患儿发病年龄分布主要集中于1岁以内,发作形式主要表现为局限性发作,早期即以婴儿痉挛起病的占21%,其中1例由局限性发作进展而来。值得注意的是,本研究中4例婴儿痉挛的患儿TSC突变基因型均为TSC2突变,推测伴有婴儿痉挛的TSC患儿可能与TSC2突变基因型相关。本研究比较了TSC1和TSC2基因突变2组患儿的癫 发生率和控制情况,发现癫
发生率和控制情况,发现癫 发生和严重程度与TSC突变基因型无明显相关性。
发生和严重程度与TSC突变基因型无明显相关性。
TSC2突变基因型、早期癫 发作和难以控制的癫
发作和难以控制的癫 是TSC患者合并智力障碍的高危因素。本研究TSC2基因突变组智力障碍发生率达85%,高于TSC1基因突变组的50%,TSC2基因突变组出现中重度智力障碍患儿亦多于TSC1基因突变组;由于本研究组样本少,Fisher's精确概率分析统计学无明显差异,但比值的结果与既往研究类似[13]。智力障碍的发生,部分原因与发育脑二次打击突变有关,而TSC患者大部分突变在TSC2基因上。本研究中92%有癫
是TSC患者合并智力障碍的高危因素。本研究TSC2基因突变组智力障碍发生率达85%,高于TSC1基因突变组的50%,TSC2基因突变组出现中重度智力障碍患儿亦多于TSC1基因突变组;由于本研究组样本少,Fisher's精确概率分析统计学无明显差异,但比值的结果与既往研究类似[13]。智力障碍的发生,部分原因与发育脑二次打击突变有关,而TSC患者大部分突变在TSC2基因上。本研究中92%有癫 患儿出现智力障碍,无癫
患儿出现智力障碍,无癫 的6号TSC2突变因孤独症表现为智力障碍,余智力发育正常。Babcock等[16]认为婴儿痉挛可作为TSC患者出现智力障碍的独立危险因素,van Eeghen等[17]也认为TSC2基因突变导致婴儿痉挛风险概率是TSC1基因突变的1.56倍,及无TSC基因突变的2.36倍,Chou和Chang[18]研究显示TSC有智力障碍与癫
的6号TSC2突变因孤独症表现为智力障碍,余智力发育正常。Babcock等[16]认为婴儿痉挛可作为TSC患者出现智力障碍的独立危险因素,van Eeghen等[17]也认为TSC2基因突变导致婴儿痉挛风险概率是TSC1基因突变的1.56倍,及无TSC基因突变的2.36倍,Chou和Chang[18]研究显示TSC有智力障碍与癫 是否控制有强相关性。另外,有研究显示有智力障碍的TSC患者更可能表现出孤独症特质,其中癫
是否控制有强相关性。另外,有研究显示有智力障碍的TSC患者更可能表现出孤独症特质,其中癫 发作是TSC患者合并孤独症倾向的重要危险因素,尤其是婴儿痉挛患者。本研究5例婴儿痉挛患儿虽然存在智力障碍,但均未见有孤独症样行为,而符合孤独症标准的6号TSC患儿无癫
发作是TSC患者合并孤独症倾向的重要危险因素,尤其是婴儿痉挛患者。本研究5例婴儿痉挛患儿虽然存在智力障碍,但均未见有孤独症样行为,而符合孤独症标准的6号TSC患儿无癫 发作,提示伴有癫
发作,提示伴有癫 的TSC患者与孤独症的关系并不清楚。
的TSC患者与孤独症的关系并不清楚。
皮质和皮质下结节、室管膜下结节和室管膜下巨细胞型星形细胞瘤是TSC主要的神经影像学改变,其发生率分别为82%~100%,83.4%~100%,5%~18%[19,20]。本研究19例TSC患儿均未见到SEGA,皮质或皮质下多发结节数目均≥3个,室管膜下结节状短T1、T2信号影仅有6例<3个,但TSC2和TSC1基因型突变与结节数量多少无明显相关性。神经系统以外的组织器官损害与TSC基因型突变的分析中,发现2、9号2例TSC2突变伴面部血管纤维瘤,其中2号背部皮肤伴有鲨鱼皮斑和肾脏错构瘤;42%TSC2突变基因型患儿眼底可见大小不等的白色团块或白斑,推测与TSC1基因突变相比,TSC2基因突变患儿更容易出现多种临床表型。
除了有联合基因缺失所产生的临床表现外,目前尚未发现某一表型与TSC某一基因型之间的对应关系[21]。本研究提示TSC2基因突变可能是我国TSC患儿主要的分子发病机制,TSC2基因发生突变的患儿其临床表型更严重,较易发生癫 、智力障碍、面部血管纤维瘤及肾错构瘤等,但尚需进一步进行多中心、大样本研究证实。
、智力障碍、面部血管纤维瘤及肾错构瘤等,但尚需进一步进行多中心、大样本研究证实。

























