
探讨尿黑酸尿症的临床特征和基因突变特点。
收集先证者及其家系成员临床资料,尿气相色谱分析尿液,采用PCR方法对尿黑酸1,2-二氧化酶(HGD)基因所有外显子检测,确定基因突变位点,Polyphen软件预测其蛋白质功能,分析基因型与表型的关系。
患者临床表现仅尿液为红褐色,而无皮肤、关节及脏器改变,尿气相色谱提示尿黑酸尿症(AKU),HGD基因检测提示该家系先证者的HGD基因第2外显子c.34A>C(p.N12H)及第4外显子c.240A>T(p.Q80H)及第12外显子c.910A>G(p.K304E)错义突变,先证者表型正常的母亲及姐姐发现携带第2外显子c.34A>C(p.N12H)错义突变,先证者表型正常的父亲发现携带第12外显子c.910A>G(p.K304E)错义突变。蛋白质功能预测后提示2号外显子c.34A>C(p.N12H)致病性突变位点。
此家系中先证者为HGD基因复合杂合子突变致病,其第2外显子c.34A>C(p.N12H)来自于母亲,第12外显子c.910A>G(p.K304E)错义突变来自于父亲,其父母及姐姐均为表型正常的杂合子携带者。发现2个国际上尚未报道的新HGD基因突变(p.N12H与p.K304E),且发病年龄最早,仅有黑尿表现。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
尿黑酸尿症(AKU)是由于尿黑酸1,2-二氧化酶(HGD)基因缺陷导致的罕见常染色体隐性遗传代谢缺陷病,全球患病率为1:(100 000~250 000),斯洛伐克发病率最高,为1:19 000[1]。江西省儿童医院内分泌遗传代谢科经尿气相色谱检查确诊AKU 1例,同时完善其家系HGD基因检测,以探讨尿黑酸尿症的临床特征和基因突变特点。
患儿,女,1月龄,因解黑尿1个月就诊。患儿母亲发现患儿解红褐色尿液,随时间变化最后变为黑色。平素体健,吃奶及生长发育良好。当地及江西省儿童医院门诊尿常规、血常规正常。肝肾功能正常。腹部(肝、胆、脾、胰)彩超未及明显异常。查体:营养中等,生长发育正常,全身皮肤色泽正常,关节无肿胀及压痛,心肺听诊无异常,肝脾无肿大,肾脏未及压痛及包块。第5胎,第2产,母亲前3胎均在孕早期不明原因自然流产;出生体质量及身长均在正常范围。父母均为汉族,家族中无黑尿家族史,否认遗传病史,否认近亲结婚。本研究获得患者家长知情同意,并签署知情同意书,同时获得医院医学伦理委员会批准。
用血串联质谱仪(MS/MS)、尿气相色谱/质谱(GC/MS)分析进行氨基酸和有机酸代谢障碍检测。
分别抽患儿及父母血3 mL,常规抽提血液基因组DNA[2]。采用Primer 5.0软件设计并合成引物。PCR反应体系50 μL,包括上游引物2.0 μL、下游引物2.0 μL、Taq酶0.4(1 U),10×Buffer 5 μL,dNTP 4.0 μL,Mg+1.0μL,ddH2O 33.6 μL,DNA(50 mg/L)2.0 μL。反应条件为95 ℃变性5 min,58 ℃退火30 s,72 ℃延伸30 s,30个循环后72 ℃延伸10 min。PCR样品用ABI遗传分析仪进行测序[3],测序结果结合人类基因突变数据库(HGMD)进行分析。
用Polyphen软件预测HGD基因突变蛋白质功能(http://genetics.bwh.harvard.edu/pph2)。
先证者为1月龄婴儿,因尿液呈黑色就诊,出生体质量3.56 kg,出生身长51 cm,未见其他阳性体征,第5胎,第2产,自然流产3胎,否认家族中类似病史。先证者、父母及姐姐完善HGD基因突变筛查,见图1。
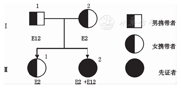
注:E:外显子;HGD:尿黑酸1,2二氧化酶 E:exons;HGD:homogentisate 1,2 dioxygenase
血液相色谱氨基酸及有机酸检测结果正常,尿气相色谱提示尿黑酸(HGA)为163.2 μmol/L(正常值0.0~1.4 μmol/L),提示AKU。
对4例家庭成员HGD基因进行测序,发现先证者父亲(Ⅰ 1)HGD基因12号外显子有一个错义突变c.910A>G(p.K304E),先证者母亲(Ⅰ 2)和姐姐(Ⅱ 1)HGD基因2号外显子上有一个错义突变c.34A>C(p.N12H)。先证者(Ⅱ 2)除了从父母分别遗传HGD基因2号和12号外显子的错义突变,还发现4号外显子(p.Q80H)错义突变,该突变与HGD基因rs2255543一致,属于编码区内的单核苷酸多态性,属于正常人群变异。先证者HGD基因2号和12号外显子错义突变暂无文献报道,见图2。

注:E:外显子;p:蛋白质;HGD:尿黑酸1,2二氧化酶 E:exon;p:protein;HGD:homogentisate 1,2 dioxygenase
用在线工具Polyphen软件对该家系HGD基因2个错义突变(p.N12H、p.K304E)进行蛋白质功能预测分析,结果表明,c.34A>C(p.N12H)对HGD蛋白质功能有致病性影响有害性风险值为0.985(0~1.000),而c.910A>G(p.K304E)有害性风险值为0.007,提示对HGD蛋白功能影响小,可能为非致病性突变。
AKU又称褐黄病,是HGD基因缺陷导致苯丙氨酸和酪氨酸的中间代谢产物HGA不能转变为延胡索酸和乙酰乙酸,致大部分HGA经肾脏排出体外,经氧化聚合转化为黑色不溶性物质,使尿液久置后变黑,故称为AKU。
HGD酶编码六聚体蛋白,2个三聚体(类似盘子样三聚体折叠成6聚体),活性部位以C端结构域和三聚体表面构成,包括His292,His335,His365,His371,and Glu341。HGA通过Fe2+与Glu341,His335和His371活性部位结合。许多氨基酸残基之间的非共价键对维持单体、三体和六聚体结构是必需的。HGD基因突变可通过以下几个方面影响HGD功能:(1)直接影响活性区域残基,干扰辅因子结合;(2)间接影响活性区域,干扰活性区域构象;(3)影响HGD亚基折叠,干扰内部疏水结构或干扰静电相互作用力,或从一个重要位置移走一个极性氨基酸,或引入不利空间联系;(4)影响内部亚基相互作用,在三体或三体之间;(5)C-端截断突变–导致内部作用消失并且正常结构缺陷。
AKU主要临床特征为尿液中存在HGA(黑尿)、褐黄病(结缔组织存在蓝黑色色素沉积)、脊柱和大关节关节炎。尿液改变通常为黑尿或将尿液加入碱性试剂后变黑,然而有些尿液放置数小时或许多个体从未观察到尿液异常。结缔组织病变通常出现在30岁以后,包括跟腱增厚、肌腱炎、疝气。褐黄病性关节炎(包括脊柱)为尿黑酸尿症常见改变[4,5]。由于HGA在肾脏代谢,肾功能受损会加重褐黄病关节炎和关节损害。其他表型有色素沉积、主动脉瓣或二尖瓣钙化或关闭不全或是主动脉扩张、肾结石、前列腺结石等。
AKU患者尿HGA水平通常为1~8 g/d(健康人24 h尿液HGA水平为20~30 mg)。AKU诊断由尿气相色谱检测出大量HGA确诊。编码尿黑酸1,2-二氧化酶的HGD基因是目前已知导致AKU的唯一基因。本例患儿出生后即有黑尿表现,为目前已报道病例中发病年龄最早,该临床表型可能与其HGD基因突变位置有关。
HGD基因位于人类染色体3q21-23,长约54 363 bp,编码445个氨基酸,由14个外显子构成。HGD基因表达只限于肝脏、肾脏、小肠、结肠和前列腺。国外报道的HGD基因致病性热点突变为外显子6、8、10、13[6,7,8,9],72.8%的突变位于氨基酸序列361~496,以错义突变最为常见(66.4%),其次是移码突变(12.2%)。Habbal等[10]报道了第1例由于HGD基因2号外显子缺失导致尿黑酸尿症。Yang等[11]报道了我国1例HGD基因突变为外显子7(p.IVS7+1G>C),外显子12(p.F329C)。胡曼等[12]发现1例HGD基因突变为外显子4(p.E74V),外显子13(p.S366-T367insTS)。Li等[13]报道了我国1例HGD基因突变为3号外显子移码突变(c.115delG)和5号外显子可变剪切。
本患儿突变位点位于第2和12外显子,与国外报道[9]热点突变位点不一致,提示我国本基因突变位点可能与其他国家存在差异。经过对2个突变位点蛋白质功能预测,提示2号外显子(p.N12H)为致病性改变,来自于母亲,但母亲及姐姐未发病,提示可能为复合杂合子(p.N12H、p.K304E)突变影响蛋白质功能,且该2个突变位点位于基因保守区和蛋白质功能区,可能为致病性变异,进一步需要完善蛋白质功能检测,明确其发病机制。
由于本病多在30岁以后出现关节、皮肤等改变,不影响生长发育。国外研究发现抗坏血酸和低蛋白饮食未发现明显疗效,目前建议定期监测并发症[14]。国外有新药尼替西农,但仍处于探索阶段[15,16]。本病的预后尚可,但可因关节和脊柱受累而致残,死因多为心血管疾病和尿毒症,成年后需随访监测心血管功能及肾功能。

























