假性甲状旁腺功能减退症(PHP)是一种罕见的遗传病,其临床特点以甲状旁腺激素抵抗为主,部分合并典型的Albright遗传性骨营养不良,表现复杂多样,易漏诊或误诊。现重点介绍PHPⅠ型的发病机制、临床表现及诊疗进展,以提高本病的诊断水平。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
假性甲状旁腺功能减退症(pseudohypoparathyroidism,PHP)是一种罕见的遗传病,于1942年由Albright等首次报道。日本学者报道其患病率约3.4/1 000 000[1]。其临床特征为甲状旁腺激素(PTH)抵抗,部分合并典型的Albright遗传性骨营养不良(Albright's Hereditary Osteodystrothy,AHO)。根据肾近曲小管对牛PTH提取物的反应,PHP分为Ⅰ型和Ⅱ型。即注射PTH后尿环磷酸腺苷等(cAMP)和尿磷排出量均不增加者,为PHP Ⅰ型;尿cAMP升高,尿磷不增加者为PHP Ⅱ型。临床PHP Ⅱ型比较少见,其发病机制尚不明确,现着重对PHP Ⅰ各型的发病机制、临床表现及诊疗进展进行综述。
根据临床表现、激活性G蛋白a亚基(a subunit of G protein,Gsa)活性及GNAS1基因缺陷类型等,PHP Ⅰ型可进一步分为Ⅰa、Ⅰ b、Ⅰc 3个亚型。PHP Ⅰc的临床表现与PHP Ⅰa相似,区别在于Ⅰc患者各种组织细胞膜上的Gsa活性正常,大部分患者未发现突变基因。
PHP Ⅰa型,多呈常染色体显性遗传,其发病与GNAS1的母源等位基因突变有关。GNAS1基因定位于20q13,由13个外显子和12个内含子组成,目前已报道193种GNAS1基因突变,包括错义突变、截断突变、剪接位点突变及小/大片段缺失或重复[2]。GNAS1基因上游不同的启动子和共同的2~13号外显子通过选择性剪接、反义转录、翻译可产生多种基因产物,其主要产物是Gsa。Gsa为cAMP/蛋白激酶A信号传导通路的重要成分,PTH主要通过PTH受体与Gsa偶联形成复合物后激活腺苷酸环化酶,促进cAMP的生成而发挥作用。Gsa活性下降,最终导致终末靶器官(肾脏、骨骼等)对PTH抵抗,临床出现钙磷代谢紊乱为特征的甲状旁腺功能减退症状,辅助检查提示低血钙、高血磷及高PTH血症。部分患者出现典型AHO畸形,即身材矮小、中心型肥胖、圆脸、短颈、指/趾粗短、异位钙化,可并智力障碍。指/趾粗短是AHO最具特征性的表现,在PHP Ⅰa中发生率为70%[3]。异位钙化在AHO患者极为常见,多见于皮下结缔组织及脑实质内,以大脑基底核钙化最常见,癫 样发作除与低钙血症相关外,也可因脑实质钙化引起。虽然PHPⅠa的临床表型与基因型无明显相关性,但指(趾)粗短、异位钙化更多见于错义突变[4],外显子7的4 bp缺失常出现AHO畸形[5]。
样发作除与低钙血症相关外,也可因脑实质钙化引起。虽然PHPⅠa的临床表型与基因型无明显相关性,但指(趾)粗短、异位钙化更多见于错义突变[4],外显子7的4 bp缺失常出现AHO畸形[5]。
母系遗传来源的内分泌腺体(如甲状腺、垂体及性腺)均与Gsa蛋白的转录有关,故GNAS1母源等位基因突变患者可出现多种激素抵抗,包括促甲状腺激素(TSH)、生长激素释放激素(GHRH)、促性腺激素。PHP Ⅰa患者均并TSH抵抗,多数为亚临床甲状腺功能低下,通常不伴甲状腺大,甲状腺相关抗体阴性[6]。少数患者新生儿期筛查即发现TSH升高。由于存在促性腺激素抵抗,患者可表现为性腺发育迟滞或性功能减退,女性患者出现月经量减少、闭经或不育,血性激素水平减低,但尚乏血促性腺激素水平升高的研究证据[7]。由于存在GHRH抵抗,矮小被公认为PHP Ⅰa患者常见的临床表现之一。但部分患者血生长激素(GH)正常,GHRH水平并不升高,提示矮小可能与骨骺过早闭合有关。部分患者胰岛β细胞功能下降,胰岛素分泌减少,并存在胰岛素抵抗,易并发糖尿病。此外,由于下丘脑黑皮素4受体通路障碍,大麻素1受体上调以及交感神经兴奋性下降,静息能量消耗减少,患者可出现早发型肥胖,进一步加重胰岛素抵抗[8]。
PHP Ⅰb型是一种罕见的基因组印记病,多呈散发,少数呈常染色体显性遗传。GNAS1基因上游的启动子区是3个差异性甲基化区域(DMRs),从5'端到3'端依次为NESP55、XLas、A/B,其中NESP55在父源等位基因发生DNA甲基化,只表达母源等位基因的拷贝,而XLas及A/B在母源等位基因发生甲基化,只表达父源等位基因的拷贝。外显子XLas与外显子2~13选择性剪接后编码一种比Gsa分子质量更大的信号蛋白,在体外可与PTH或其他内分泌激素受体结合,在转基因鼠的肾近曲小管可增强Gsa的作用,但在人体内的功能仍未明确[9]。外显子A/B具有调控Gsa组织特异性父系表达的顺势作用元件。常染色体显性遗传的PHP Ⅰb(AD-PHPⅠb)常为外显子A/B的甲基化缺失引起[10]。而A/B上游区域的突触融合蛋白-16(STX-16)包含与A/B甲基化相关的顺式作用元件[11],因此STX-16基因微缺失与A/B甲基化缺陷相关,其中以3.0 kb微缺失最常见,也可见4.4 kb及24.6 kb的基因片段缺失[12]。NESP55的母源等位基因片段缺失也可见于AD-PHP Ⅰb,若突变位于父源等位基因,携带者可不发病。散发PHP Ⅰb常涉及多个DMRs,如20q父源单亲二倍体,即使不携带任何突变基因,仍可出现广泛的甲基化异常[13]。PHP Ⅰb与GNAS1基因的表遗传调控异常有关,编码基因序列未发生改变,患者红细胞膜或成纤维细胞膜上的Gsa活性正常。其临床表现除PTH抵抗外,多不合并其他激素抵抗,但有报道少部分患者存在轻度TSH抵抗[14]。有研究显示PTH抵抗的轻重与甲基化缺失的程度相关[15]。PHP Ⅰb多不伴AHO畸形,但部分患者可出现指/趾粗短[16]。
此外,假假性甲状旁腺功能减退症(pseudo-pseudohypoparathyroidism,PPHP)是一类具有AHO特殊体型,但无PTH抵抗的综合征,由GNAS1的父源等位基因突变所致。PPHP患者在胎儿时期可出现严重的宫内发育迟缓,提示GNAS1基因突变与胎儿宫内迟缓相关,且父系遗传比母系遗传的更严重,外显子XLas与胎儿的宫内发育有关[17]。而AHO的常见症状,如肥胖、认知障碍更常见于PHP Ⅰa,这一现象与Gsa在鼠的脂肪及脑细胞中以母系表达为主的特点相吻合[18]。各型PHP患者的特点可总结见表1。
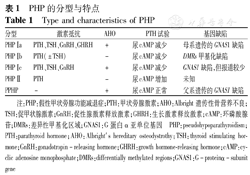
PHP的分型与特点
Type and characteristics of PHP
PHP的分型与特点
Type and characteristics of PHP
| 分型 | 激素抵抗 | AHO | PTH试验 | 基因缺陷 |
|---|---|---|---|---|
| PHP Ⅰa | PTH、TSH、GnRH、GHRH | + | 尿cAMP减少 | 母系遗传的GNAS1缺陷 |
| PHP Ⅰb | PTH(±TSH) | - | 尿cAMP减少 | DMRs甲基化缺陷 |
| PHP Ⅰc | PTH、TSH、GnRH | + | 尿cAMP减少 | GNAS1缺陷,但报道较少 |
| PHP Ⅱ | PTH | - | 尿cAMP增加 | 未知 |
| PPHP | - | + | 尿cAMP正常 | 父系遗传的GNAS1缺陷 |
注:PHP:假性甲状旁腺功能减退症;PTH:甲状旁腺激素;AHO:Albright遗传性骨营养不良;TSH:促甲状腺激素;GnRH:促性腺激素释放激素;GHRH:生长激素释放激素;cAMP:环磷酸腺苷;DMRs:差异性甲基化区域;GNAS1:G蛋白α亚单位基因 PHP:pseudohypoparathyroidism;PTH:parathyroid hormone;AHO:Albright's hereditary osteodystrothy;TSH:thyroid stimulating hormone;GnRH:gonadotropin-releasing hormone;GHRH:growth hormone-releasing hormone;cAMP:cyclic adenosine monophosphate;DMRs:differentially methylated regions;GNAS1:G-proteinα-subunit gene
随着对本病研究的深入,更为复杂的基因型和临床表型得到揭示,既往的分型方法面临很大争议和挑战,例如:(1)PHP Ⅰa及PPHP患者均有AHO畸形及Gsa活性减低的特点,但20%~30%的患者并未发现GNAS1基因的致病突变,其发病机制仍未阐明。(2)PHP Ⅰa与PHP Ⅰc的区别在于Gsa活性是否正常,后者一般无母系来源的GNAS1缺陷,但也有研究在PHP Ⅰc患者(AHO合并PTH抵抗,Gsa活性正常)中发现GNAS1基因存在致病性突变[19]。这些Ⅰc型患者在临床、生化及基因水平上与Ⅰa型无任何差异。(3)有报道临床表现为PHP Ⅰa(AHO合并PTH抵抗)的患者,基因分析却发现与PHP Ⅰb相关的基因缺陷,即GNAS1上游DMRs区域存在缺陷[3]。(4)Turan等[20]发现1例父系遗传的外显子A/B区的新发突变c.328 G>C,该患者确诊为PPHP,但其具有PTH抵抗的生化改变。可见,目前的分类可能存在临床、生化、分子及基因水平上的重叠。由于分型并不一定能够指导治疗及判断预后,对于特征不典型的患者不强调分型。
PHP可在任何年龄发病,若已出现特征性症状、体征及生化改变,诊断并不困难。典型临床表现有AHO畸形以及反复发作的手足搐搦、感觉异常、癫 样发作等,生化改变提示PTH抵抗。手足X线片可出现掌骨、指骨、跖骨和趾骨对称性不成比例缩短,尤以第4、5掌骨变短为特征性改变。头颅影像学检查可发现颅内多发钙化,特征性表现为基底核区"倒八字"征和大脑皮质下区对称性"星火样"钙化灶。如果患者有以上改变,并能除外原发性甲状旁腺功能减低症以及慢性肾衰竭和低镁血症所引起的高PTH血症等疾病,即可临床诊断PHP。
样发作等,生化改变提示PTH抵抗。手足X线片可出现掌骨、指骨、跖骨和趾骨对称性不成比例缩短,尤以第4、5掌骨变短为特征性改变。头颅影像学检查可发现颅内多发钙化,特征性表现为基底核区"倒八字"征和大脑皮质下区对称性"星火样"钙化灶。如果患者有以上改变,并能除外原发性甲状旁腺功能减低症以及慢性肾衰竭和低镁血症所引起的高PTH血症等疾病,即可临床诊断PHP。
对于AHO畸形不明显的病例,或疾病早期尚未出现典型PTH抵抗的生化改变时,PHP的诊断比较困难。对于这些患者,可行GNAS1基因测序。传统的GNAS1基因测序方法可以检测外显子1~13上的任意突变,从而使部分PHP Ⅰa及PPHP患者得以确诊。通过验证父母双方的GNAS1基因,可以确定突变基因的来源,父系或母系,可用于遗传咨询和下一胎的产前诊断。对于双亲之一的标本不易获取的患者,可通过反转录-PCR的方法,利用突变位点与外显子NESP55及XLas、A/B的关系,明确突变来源[20]。对于PHP Ⅰb患者需要进行DMRs区域的甲基化分析。GNAS1基因印迹分析,包括Southern印迹分析,通过检测DMRs区的胞嘧啶鸟嘌呤磷酸化位点的甲基化程度来定性或半定量反应该基因的甲基化程度。多重连接探针扩增技术(MS-MLPA)是最准确、最高效检测甲基化程度的方法。
部分PHP患者尚未发现致病基因,基因测序阴性,必要时可测定Gsa的生物活性并进行Ellsworth-Howard试验。有研究显示皮下注射重组型PTH可验证是否存在PTH抵抗,其结果与静脉注射天然的PTH一致[21]。目前测定Gsa的活性,包括半定量的免疫印迹及Gsa功能分析,但仍处于实验阶段。PHP Ⅰa及PPHP为GNAS1基因突变所致,Gsa的生物活性至少降低50%;PHP Ⅰb与DMRs的甲基化缺陷相关,大部分患者细胞膜上的Gsa活性是正常的,但有研究发现合并手指粗短的PHP Ⅰb患者Gsa活性轻度下降,提示AHO畸形可能与Gsa的生物活性相关[21]。
PHP为基因遗传性疾病,目前尚无根治方法,临床只能对症治疗,需要终生口服钙剂和活性维生素D,减少磷的摄入,维持血钙、血磷在正常水平,缓解症状。
患者出现低血钙性惊厥时应立即静脉输注钙剂,尽快控制抽搐发作,避免昏迷、喉痉挛等严重合并症。长期口服钙剂的患者,应注意监测尿钙,若出现高钙尿症,必要时可口服噻嗪类利尿剂。若患者仅口服维生素D即可维持正常的血钙、血磷,则勿需同时口服钙剂。由于长期的高PTH血症可能诱发骨折、骨畸形甚至散发性甲状旁腺功能亢进症。因此,即使不伴低钙血症,高PTH血症患者仍应给予维生素D治疗。少部分由于颅内异位钙化引发癫 者,需要抗癫
者,需要抗癫 治疗,但应避免给予苯妥英钠、苯巴比妥以及卡马西平等。
治疗,但应避免给予苯妥英钠、苯巴比妥以及卡马西平等。
PHP患者可存在多种激素抵抗,必要时应给予相应的激素替代治疗。如甲状腺功能减退者可给予L-甲状腺素;性功能发育延迟、月经量减少者给予性激素替代治疗。本病的身材矮小,除与GHRH抵抗所致GH缺乏相关外,尚与骨骺过早闭合有关,故并非均需GH替代治疗,应尽早行GH激发试验,寻找GH缺乏的证据,必要时尽早(青春期前)给予GH治疗。
AHO畸形,包括指/趾粗短等,同样缺乏有效的治疗方法。对于少数发生在特殊部位的异位钙化,如皮下结缔组织或关节附近,钙化体积较大影响外观或功能时,可通过手术移除。PHP Ⅰa患者可出现早发型肥胖,Al-Sarameh等[23]应用大麻素受体Ⅰ拮抗剂可将患者的体质量指数从40.5降至30.5,但该类药物可影响儿童的脑发育,因此临床上并不推荐。近期研究显示,PHP的肥胖患者较其他肥胖患者更容易发生睡眠呼吸暂停,发生率分别为45%和1%~5%[24]。呼吸暂停可加重认知障碍,加重代谢紊乱,增加心血管疾病的发生率。尽早干预睡眠呼吸暂停可改善PHP患者的远期预后。GH可促进脂肪代谢,一定程度上减轻肥胖,减少呼吸暂停的发生率,必要时在应用GH前可使用双相呼吸道正压辅助通气[25]。
PHP的发病机制复杂,临床表型多样化,部分患者早期症状不典型,诊断困难,即使获得诊断,目前仅能给予对症治疗。通过多项研究已经明确了GNAS1基因的表达方式、功能以及调节机制,各种新的致病基因的发现,进一步加深了对本病的认识,明确了致病突变的遗传方式,有助于产前咨询,目前有研究通过种植前基因诊断技术,使PHP患者有希望孕育健康的下一代。


























