
分析6例GM1神经节苷脂累积病患儿临床及遗传学特点。
收集2014年4月至2015年8月在北京大学第一医院儿科就诊的6例GM1神经节苷脂累积病先证者及其家系成员临床资料,并进行白细胞溶酶体酶活性检测,分析临床特点;进行β-半乳糖苷酶-1(GLB1)基因突变检测,分析遗传学特点。
1.临床特点:6例患儿均于出生8个月内起病。患儿均有智力运动发育迟缓伴倒退,多数患儿有惊跳反应、癫痫史、肌张力低下,少数患儿有视听力损害、Mongolian斑、肝大及体格生长落后。头颅磁共振成像(MRI)均表现髓鞘形成不良,4例见T2像丘脑低信号,4例见大脑和/或小脑萎缩。患儿溶酶体酶活性检测均示酸性GLB活性降低。6例患儿均符合GM1神经节苷脂累积病诊断,其中Ⅰ型3例,Ⅱ型3例。2.遗传学特点:6例患儿共发现10种GLB1突变:c.520T>C(p.Y174H)、c.622C>T(p.R208C)、c.550C>T(p.Q184X)、c.446C>T(p.S149F)、c.266 A>G(p.H89R)、c.601C>T(p.R201C)、c.148T>A(p.Y50N)、c.618delC(p.R208AfsX21)、c.1343A>T(p.D448V)、c.410C>A(p.A137D),其中c.550C>T(p.Q184X)、c.148T>A(p.Y50N)、c.618delC(p.R208AfsX21)、c.266A>G(p.H89R)及c.410C>A(p.A137D)为未报道新突变。4例患儿为复合杂合突变,2例为纯合突变,均分别遗传自表型正常的父母。
本研究确诊6例GM1神经节苷脂累积病患儿,发现5种GLB1未报道新突变,扩大了GLB1突变谱,为患儿家庭提供准确的遗传咨询及产前诊断。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
GM1神经节苷脂累积病是一种常染色体隐性遗传病,国外报道的发病率为1/20万~1/10万活产婴儿[1]。多于婴儿期或儿童期起病,临床表现包括智力运动发育迟缓伴倒退、肝脾大、眼底樱桃红斑、粗陋面容、多发性成骨不良,头颅磁共振成像(MRI)可见髓鞘化落后、丘脑及基底核异常信号,患儿酸性β-半乳糖苷酶(GLB)活性明显降低[2]。致病基因为定位于3号染色体的β-半乳糖苷酶-1(GLB1)基因。GLB1突变可导致GLB缺陷,其底物GM1神经节苷脂及相关糖复合物过量沉积于全身组织尤其是神经系统,引发细胞功能障碍、神经元应激反应、凋亡、神经炎症反应及髓鞘病变[1,3]。该病尚无统一诊断标准。GM1神经节苷脂累积病具有明显的临床及遗传学异质性,且与GM2神经节苷脂累积病、黏多糖病等其他溶酶体病表现相似,增加了诊断难度[3,4,5,6,7]。本研究拟对2014年4月至2015年8月在北京大学第一医院儿科就诊的6例GM1神经节苷脂累积病家系进行临床及GLB1突变分析,明确其临床及遗传学特点,为患儿家庭提供准确的遗传咨询及产前诊断。
入选对象为2014年4月至2015年8月在北京大学第一医院儿科就诊的6例(P1~P6)GM1神经节苷脂累积病患儿,男女各3例,年龄1岁8个月~4岁1个月,父母均身体健康,均非近亲婚配,家族成员均否认类似疾病史。入选标准参照Brunetti-Pierri和Scaglia[8]于2008年报道的GM1神经节苷脂累积病各型特点。本研究经北京大学第一医院医学伦理委员会批准,患儿法定监护人均签署知情同意书。
收集患儿及家系临床资料包括病史、体格检查及辅助检查结果。
采集患儿外周静脉血3~4 mL,委托德易东方转化医学研究中心进行溶酶体酶分析。GLB的活性通过测定4-甲基伞形酮-β-D-半乳糖苷经过白细胞GLB水解后得到的荧光产物4-甲基伞形酮进行定量。
采集先证者及家系成员外周血标本,盐析法提取白细胞基因组DNA。根据从人类基因组数据库GenBank(http://www.ncbi.nlm.nih.gov/genbank/)中获得的GLB1序列(NM_000404),使用Primer Premier 5.0软件设计引物对GLB1编码区进行PCR反应并对反应产物进行Sanger测序。结果经比对后确定核苷酸及编码氨基酸改变位点,分析家系传递特点,检索HGMD(Human Gene Mutation Database)数据库确定是否已报道;若未经报道,则通过检测50个健康人相同位点推测是否为单核苷酸多态性(Single nucleotide polymorphisms,SNP),并通过氨基酸保守性(http://genome.ucsc.edu/)、性质分析及软件(Mutationtaster,Polyphen-2)预测是否为致病性突变[9,10]。
P1~P3于出生7~8个月起病,P4~P6于出生3个月内起病。P5生前即表现宫内发育迟缓,其余患儿均以发育里程碑延迟为首发症状。智力运动发育迟缓伴倒退见于所有患儿,P1~P3在1岁4个月~1岁6个月开始倒退,P4~P6为7~9个月。P2有眼震。P4在1岁6个月对外界声光刺激已无回应,余患儿常因声音诱发惊跳反应。4例(P2~P3、P5~P6)有癫痫史。患儿均出现饮水呛咳伴咀嚼功能减退导致喂养困难。6例患儿除P5外出生史及体格生长均无明显特殊,P5足月顺产,出生体质量2.25 kg,1岁1个月时头围43.5 cm,2岁6个月时身长85 cm,体质量11.5 kg。患儿均未见粗陋面容。6例仅P5行眼底检查,未见樱桃红斑。P4可见全身Mongolian斑及肝脏进行性肿大。5例(P1~P2、P4~P6)肌张力低,P3肌张力高,P3、P5肌张力不全(表1)。6例患儿仅P4于6月龄行X线骨关节检查,未发现骨骼畸形。6例患儿头颅MRI均见髓鞘化落后,4例(P2~P3、P5~P6)见T2加权像丘脑低信号,4例(P2~P4、P6)见大脑或小脑萎缩(图1)。
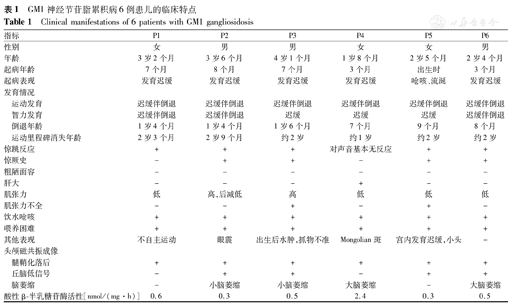
GM1神经节苷脂累积病6例患儿的临床特点
Clinical manifestations of 6 patients with GM1 gangliosidosis
GM1神经节苷脂累积病6例患儿的临床特点
Clinical manifestations of 6 patients with GM1 gangliosidosis
| 指标 | P1 | P2 | P3 | P4 | P5 | P6 | |
|---|---|---|---|---|---|---|---|
| 性别 | 女 | 男 | 男 | 女 | 女 | 男 | |
| 年龄 | 3岁2个月 | 3岁6个月 | 4岁1个月 | 1岁8个月 | 2岁5个月 | 2岁4个月 | |
| 起病年龄 | 7个月 | 8个月 | 7个月 | 3个月 | 出生时 | 3个月 | |
| 起病表现 | 发育迟缓 | 发育迟缓 | 发育迟缓 | 发育迟缓 | 呛咳、流涎 | 发育迟缓 | |
| 发育情况 | |||||||
| 运动发育 | 迟缓伴倒退 | 迟缓伴倒退 | 迟缓伴倒退 | 迟缓伴倒退 | 迟缓伴倒退 | 迟缓伴倒退 | |
| 智力发育 | 迟缓伴倒退 | 迟缓伴倒退 | 迟缓 | 迟缓 | 迟缓 | 迟缓伴倒退 | |
| 倒退年龄 | 1岁4个月 | 1岁4个月 | 1岁6个月 | 7个月 | 9个月 | 8个月 | |
| 运动里程碑消失年龄 | 2岁3个月 | 2岁9个月 | 约2岁 | 约1岁 | 约2岁 | 约2岁 | |
| 惊跳反应 | + | + | + | 对声音基本无反应 | + | + | |
| 惊厥史 | - | + | + | - | + | + | |
| 粗陋面容 | - | - | - | - | - | - | |
| 肝大 | - | - | - | + | - | - | |
| 肌张力 | 低 | 高,后减低 | 高 | 低 | 低 | 低 | |
| 肌张力不全 | - | - | + | - | + | - | |
| 饮水呛咳 | + | + | + | + | + | + | |
| 喂养困难 | + | + | + | + | + | + | |
| 其他表现 | 不自主运动 | 眼震 | 出生后水肿,抓物不准 | Mongolian斑 | 宫内发育迟缓,小头 | - | |
| 头颅磁共振成像 | |||||||
| 髓鞘化落后 | + | + | + | + | + | + | |
| 丘脑低信号 | - | + | + | - | + | + | |
| 脑萎缩 | - | 小脑萎缩 | 小脑萎缩 | 大脑萎缩 | - | 大脑萎缩 | |
| 酸性β-半乳糖苷酶活性[nmol/(mg·h)] | 0.6 | 0.3 | 0.5 | 2.4 | 0.3 | 0.5 | |
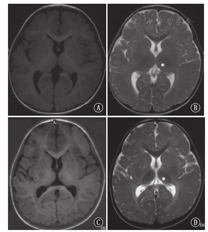
注:A:1岁6个月,T1加权像;B:1岁6个月,T2加权像;C:2岁8个月,T1加权像;D:2岁8个月,T2加权像
A:1 year and 6 months,T1W image;B:1 year and 6 months,T2W image;C:2 years and 8 months,T1W image;D:2 years and 8 months,T2W image
患儿酸性GLB活性检测值均明显降低,5例患儿(P1~P3、P5、P6)为0.3~0.6 nmol/(mg·h)[参考值12.6~52.7 nmol/(mg·h)];P4患儿为2.4 nmol/(mg·h)[参考值为50.0~140.0 nmol/(mg·h)](表1)。
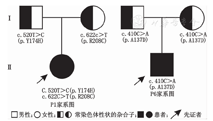
6例患儿共检测出10种GLB1突变,其中错义突变8种:c.622C>T(p.R208C)、c.520T>C(p.Y174H)、c.446C>T(p.S149F)、c.266 A>G(p.H89R)、c.601 C>T(p.R201C)、c.148T>A(p.Y50N)、c.1343A>T(p.D448V)及c.410C>A(p.A137D);截断突变1种:c.550C>T(p.Q184X);移码突变1种:c.618delC(p.R208AfsX21)。有5种突变未报道:c.550C>T(p.Q184X)、c.266A>G(p.H89R)、c.148T>A(p.Y50N)、c.618delC(p.R208AfsX21)及c.410C>A(p.A137D)。4例(P1~P4)为复合杂合突变,2例(P5~P6)为纯合突变,均分别遗传自表型正常父母(表2,图2、图3)。
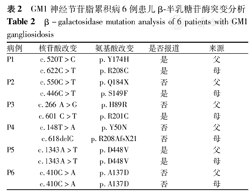
GM1神经节苷脂累积病6例患儿β-半乳糖苷酶突变分析
β-galactosidase mutation analysis of 6 patients with GM1 gangliosidosis
GM1神经节苷脂累积病6例患儿β-半乳糖苷酶突变分析
β-galactosidase mutation analysis of 6 patients with GM1 gangliosidosis
| 病例 | 核苷酸改变 | 氨基酸改变 | 是否报道 | 来源 |
|---|---|---|---|---|
| P1 | c.520T>C | p.Y174H | 是 | 父 |
| c.622C>T | p.R208C | 是 | 母 | |
| P2 | c.550C>T | p.Q184X | 否 | 父 |
| c.446C>T | p.S149F | 是 | 母 | |
| P3 | c.266 A>G | p.H89R | 否 | 父 |
| c.601 C>T | p.R201C | 是 | 母 | |
| P4 | c.148T>A | p.Y50N | 否 | 父 |
| c.618delC | p.R208AfsX21 | 否 | 母 | |
| P5 | c.1343A>T | p.D448V | 是 | 父 |
| c.1343A>T | p.D448V | 是 | 母 | |
| P6 | c.410C>A | p.A137D | 否 | 父 |
| c.410C>A | p.A137D | 否 | 母 |
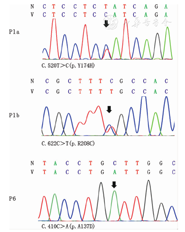
P1 and P6′s results of Sanger sequencing of GLB1
注:N:正常序列;V:变异序列;GLB1:β-半乳糖苷酶-1;箭头示GLB1突变位置;P1a/b:P1 GLB1核苷酸改变,P6:P6 GLB1核苷酸改变
N:normal sequence;V:variation sequence;GLB1:galactosidase-1;arrow:the position of nucleotide variation;P1a/b:GLB1 nucleotide variation of P1;P6:GLB1 nucleotide variation of P6
P1 and P6′s results of Sanger sequencing of GLB1
GM1神经节苷脂累积病是一组具有临床及遗传异质性的常染色体隐性遗传的溶酶体病,根据临床表现可分为3型:Ⅰ型(婴儿型,MIM#230500),最常见,出生后6月内起病,患儿寿命1~2岁,主要表现智力运动倒退、肌张力低下、粗陋面容、肝脾大、成骨不良及眼底樱桃红斑;Ⅱ型(晚发婴儿型或青少年型,MIM#230600),7个月~3岁起病,患儿主要表现智力运动倒退、成骨不良、粗陋面容及肌张力低下等;Ⅲ型(成人型,MIM#230650)于3~30岁起病,患儿多有骨骼畸形、锥体外系症状及异常步态。部分患儿可表现Mongolian斑、癫痫及心肌病。外周血淋巴细胞空泡形成、异常头颅MRI及白细胞酸性GLB活性降低对诊断起着重要作用[1,8,11,12,13,14]。
本组病例中P1~P3于7~8个月龄起病,P4~P6于出生后3个月内起病。患儿均有智力运动发育迟缓及倒退,部分病例有惊跳反应、肌张力低下、Mongolian斑、肝脾大、宫内生长迟缓、眼震等,头颅MRI均示髓鞘形成不良,GLB活性均显著降低,临床符合GM1神经节苷脂累积病诊断,P1~P3及P4~P6的发病年龄分别符合Ⅱ型及Ⅰ型。本组患儿均未见粗陋面容,可能和该病临床异质性有关。本研究6例患儿多数未能行骨关节X线及眼底检查以明确骨骼畸形及眼底樱桃红斑。
GM1神经节苷脂累积病头颅MRI可表现为大脑及小脑萎缩;白质T2加权像高信号,提示髓鞘形成不良;胼胝体发育不良;基底核及苍白球T2加权像低信号;壳核T2加权像高信号[15,16,17,18]。本组病例头颅MRI均可见髓鞘形成不良,部分病例可见脑萎缩、基底核或丘脑异常信号,符合GM1神经节苷脂累积病头颅MRI表现。
GLB活性检测对GM1神经节苷脂累积病诊断起重要作用。通过4-甲基伞形酮-β-D-半乳糖法检测的Ⅰ型、Ⅱ型及Ⅲ型患者成纤维细胞GLB活性分别为正常值的0.07%~1.30%、0.07%~1.30%及9.00%。需要注意的是,半乳糖唾液酸沉积症亦可表现GLB活性降低,但后者并存α-神经氨酸苷酶活性降低,临床表现以角膜云翳、骨骼发育畸形为主,无明显神经系统症状[1,8]。
GLB1位于染色体3p21.33,全长62.5 kb,包含16个外显子,编码长度为667个氨基酸的GLB及546个氨基酸的弹性蛋白结合蛋白(EBP)。GLB是细胞溶酶体酶,可水解神经节苷脂、糖蛋白及黏多糖的β-半乳糖苷基团;EBP则通过与细胞保护性蛋白/组织蛋白酶A(PPCA)及神经氨酸酶(NEU1)形成复合物,参与细胞膜表面弹性纤维的形成。GLB1突变可导致GM1神经节苷脂累积病或黏多糖病ⅣB型(MIM#253010),可能与突变的位置不同有关。当突变位于GLB1蛋白的核心桶装作用域时可导致GM1神经节苷脂累积病,当突变位于GLB和EBP的共同编码区或配体结合区域时可导致黏多糖病ⅣB型[1,8,19]。导致GM1神经节苷脂累积病的GLB1突变超过150种,包括点突变(>99%)、无义突变、移码突变、重复及缺失突变,其中p.R482H、p.R208C、p.R201C及p.I51T为热点突变[1,8,20]。本组6例患儿共检测出10种GLB1突变,其中热点突变2种:c.622C>T(p.R208C)及c.601G>A(p.R201C),未报道突变5种:c.266T>C(p.H89R)、c.148T>A(p.Y50N)、c.410C>A(p.A137D)、c.550C>T(p.Q184X)及c.618delC(p.R208AfsX21)。
P1发现GLB1 c.520T>C(p.Y174H)及c.622C>T(p.R208C)改变。p.Y174H已报道于Ⅱ型GM1神经节苷脂累积病[15];p.R208C在3型中均有报道(http://www.ncbi.nlm.nih.gov/clinvar/),患儿p.Y174H来自表型正常父亲,p.R208C来自表型正常母亲,据此推测P1为GLB1 c.520T>C(p.Y174H)及c.622C>T(p.R208C)复合杂合突变致病。P2发现GLB1 c.550C>T(p.Q184X)及c.446C>T(p.S149F)改变。患儿p.Q184X改变导致蛋白翻译终止,来自表型正常父亲;患儿p.S149F报道于GM1神经节苷脂累积病Ⅲ型[21],来自正常母亲,可推测P2为GLB1 c.550C>T(p.Q184X)及c.446C>T(p.S149F)复合杂合突变。P3发现GLB1 c.266T>C(p.H89R)及c.601G>A(p.R201C)改变。患儿p.H89R来自正常父亲,其导致氨基酸性质改变,非SNP,氨基酸位点高度保守,经预测具有致病性;患儿p.R201C来自母亲,该突变曾报道于GM1神经节苷脂累积病Ⅱ型(http://www.ncbi.nlm.nih.gov/clinvar/)。据此推测P3为GLB1 c.266T>C(p.H89R)及c.601G>A(p.R201C)复合杂合突变。P4发现GLB1 c.148T>A(p.Y50N)及c.618delC(p.R208AfsX21)改变,分别来自表型正常父母。p.Y50N导致氨基酸性质改变;p.R208AfsX21导致p.R208氨基酸改变,其后第21个密码子变为终止密码。p.Y50N及p.R208AfsX21均非SNP改变,氨基酸位点高度保守,经预测具有致病性,推测P4为GLB1 c.148T>A(p.Y50N)及c.618delC(p.R208AfsX21)复合杂合突变。P5发现GLB1 c.1343A>T(p.D448V)改变,分别来自父母。该突变已报道[22],并有研究发现在转染p.D448V的过表达GLB的COS-1细胞中GLB活性几乎为0[16],说明有致病性,可推测P5为GLB1 c.1343A>T(p.D448V)纯合突变。P6发现GLB1 c.410C>A(p.A137D)改变,同样分别来自父母。该改变可改变氨基酸性质,此外,p.A137相邻位点p.P136在GLB1蛋白内部核心区域,p.P136S突变可导致GM1神经节苷脂累积病Ⅰ型[19]。c.410C>A非SNP改变,氨基酸位点高度保守,经预测具有致病性,故推测P6为c.410C>A(p.A137D)纯合突变致病。综上所述,本组6例患儿均为GLB1突变致病,其中4例(P1~P4)为复合杂合突变,2例(P5~P6)纯合突变。
本研究通过白细胞溶酶体酶检测及GLB1突变检测,确诊3例Ⅰ型及3例Ⅱ型GM1神经节苷脂累积病,发现5种GLB1未报道新突变,扩大了GLB1突变谱。GM1神经节苷脂累积病具有明显的临床及遗传学异质性,本组患儿均以智力运动发育迟缓伴倒退为主要表现,无粗陋面容、少见肝脾大等典型表现。GLB活性降低及GLB1突变检出对于不典型GM1神经节苷脂累积病诊断有重要意义。此外,GLB1突变检出也为患者家庭的遗传咨询及产前诊断提供了可能。通过进一步的大样本基因突变分析,将可能得出我国GM1神经节苷脂累积病致病基因突变谱,这将不仅有助于将来在我国开展快速的GLB1突变检测,而且能够为深入研究该基因突变的功能特性、探讨其发病机制并寻找新的治疗方法打下坚实的基础。

























