
我国自1981年起,逐步开展了高苯丙氨酸血症(HPA)的新生儿筛查,许多苯丙酮尿症(PKU)患儿生长发育良好,一些患儿已步入婚育阶段。女性患者孕期血苯丙氨酸(Phe)浓度持续增高,可导致胎儿脑发育障碍及各种畸形发生,即母源性PKU综合征。现从母源性PKU的发生机制、预防、四氢生物蝶呤的支持治疗及成年女性PKU患者的综合管理等各个方面,综述国际上的最新进展,以期对于女性PKU患者顺利度过孕产期,保证胎儿的健康成长,防止母源性PKU的发生提供一些借鉴。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
高苯丙氨酸血症(hyperphenylalaninemia,HPA)是我国最常见的先天性氨基酸代谢病,为常染色体隐性遗传病,是第1个可治疗的遗传病,通过早期诊断、早期治疗可避免患儿发生智力残疾,为全球新生儿筛查的首选病种。患者的主要生化特征为血苯丙氨酸(phenylalaninine,Phe)浓度持续增高超过120 μmol/L,血Phe与酪氨酸(tyrosine,Tyr)比值>2.0。HPA分为2种类型:一种是由于肝脏苯丙氨酸羟化酶(phenylalanine hydroxylase,PAH)基因突变,PAH活性降低或丧失,食物中的Phe无法转化为酪氨酸,Phe及其衍生物在体内蓄积,旁路代谢增强,大量苯丙酮酸、苯乙酸和苯乳酸从尿中排出,又称为苯丙酮尿症(phenylketonuria,PKU);另一种类型是PAH的辅酶——四氢生物蝶呤(tetrahydrobiopterin,BH4)合成或代谢途径中某种酶的先天缺陷引起的BH4缺乏症。2种类型疾病的治疗方法不同,PKU的治疗方法主要为低Phe饮食,而BH4缺乏症的治疗主要为BH4等药物支持,故早期鉴别诊断是指导治疗的关键[1,2]。
体内高浓度的Phe及其旁路代谢产物损伤患者的神经系统,如不能早期干预,最终导致患者智力残疾。控制Phe的摄入或针对BH4缺乏症进行相应的治疗可有效防止病理损害的发生。各个国家与地区HPA的发病率及疾病谱有所不同。我国1985年至2011年3 500万新生儿筛查资料显示,HPA患病率为110 397,其中87.1%以上为PAH基因缺陷所致,12.9%为BH4缺乏症,以6-丙酮酰四氢蝶呤合成酶(6-pyruvoyl-tetrahydropterin synthase,PTPS)缺乏最常见,并存在显著的地域差异,南部地区BH4缺乏症发病率较高[1]。
HPA患者新生儿期多无临床表现。出生1~2个月逐渐出现异常,头发由黑变黄,皮肤颜色浅淡,尿液汗液鼠尿味,随年龄增长,智力发育落后明显,小头畸形,癫痫发作,也可出现行为、性格、精神异常,如多动、智残、攻击、自闭、自卑、抑郁等。自1981年起,我国逐步开展了HPA的新生儿筛查,越来越多的PKU患儿在无症状时期获得诊断与治疗,获得了良好的智能和体格发育,其中一些患儿已经成年,进入婚育阶段。如疾病控制不良,持续的HPA对精子、卵子及胚胎毒性很强。尤其是女性患者孕期血Phe浓度持续增高,可导致胎儿脑发育障碍及各种畸形发生,即母源性PKU综合征。尤其是女性患者,如何顺利度过孕产期,保证胎儿的健康成长,防止母源性PKU的发生,已成为重要课题。对PKU女性患者需进行产前咨询,在孕前6个月至整个孕期需要饮食治疗,控制血Phe在120~360 μmol/L[1]。
低Phe饮食治疗仍是目前PAH缺乏症的主要治疗方法。PKU患者PAH酶活性不同,对Phe的耐受量个体差异显著,需要终生的个体化治疗。现实中约75%的青少年及成年PKU患者治疗依从性较差,中断治疗或血Phe控制不理想者仍会导致一系列精神、行为等异常[1],需要坚持治疗。由于患者及其家人理解不足,缺乏保险和财政支撑,患者认知、精神、神经功能受损等多方面因素,加之饮食治疗使用的低Phe食品口感不好,成年患者对特殊饮食的依从性较差。国内外的现状是6岁以后很多患者放松了饮食控制,很多成年患者,尤其是孕妇,血Phe浓度未得到良好的控制。
孕妇体内的血Phe通过主动转运送达胎盘,胎儿血Phe浓度可高于母体1.5~2.0倍。持续过高浓度的血Phe对胎儿体内各脏器的发育产生毒性,尤其是对胎儿脑和心脏损害严重。有文献报道,血Phe浓度控制不良PKU孕母产下的婴儿,最常见的异常包括宫内及出生后发育迟缓、小头畸形、先天性心脏缺损、颅面骨畸形、面容异常,脊柱发育异常、斜视,甚至出现腭裂、膀胱外翻等严重畸形[3]。血Phe浓度控制不良的PKU孕妇所生的婴儿92%出现发育迟缓,73%出现小头畸形,12%出现先天性心脏病伴有发育迟缓及癫痫[4]。胎儿中枢神经系统、心脏等重要脏器发育的关键时期是妊娠早期第5-8周,如果在此之前,不能控制母体血Phe浓度,高浓度血Phe会通过胎盘进入胎儿血循环,导致畸形。
Rouse等[5]研究证实,随着孕母血Phe浓度的增高,婴儿的先天异常发生率也相应增高。高血Phe浓度的孕母,小头畸形婴儿的发生率高达85%。如果孕母从怀孕前开始,并在整个怀孕期,血Phe浓度均维持在120~360 μmol/L,小头畸形婴儿的出生率可降到3%。Widaman和Azen[6]也报道,如果孕母的血Phe浓度维持在600 μmol/L以下,小头畸形的发病率就会从73%降至8%。Grange等[7]观察了21例PKU孕妇血Phe控制情况,其中5例在怀孕期间血Phe浓度经常高于360 μmol/L,2例流产,1例胎儿腭裂,正常出生婴儿比例为2/5例,占40%;另外16例血Phe浓度常低于360 μmol/L,其中2例自然流产,2例早产,正常婴儿出生比例为12/16例,占75%。
Waisbren等[8]曾对182例PKU母亲所生的儿童进行了随访研究,4岁时进行智力测试,结果证实儿童的智商与其母亲的血Phe浓度成反比。如果母亲在怀孕20周内未控制血Phe浓度,所生子代47%智商会低于正常值的2个标准差(SD)以下,而在怀孕前就开始控制血Phe浓度的,子代智商正常。
Moseley等[9]还报道了2例PKU孕妇,智商分别为113分和108分,均接受了大学教育,妊娠期间血Phe浓度控制在360 μmol/L,所生婴儿发育正常。1例是经过新生儿筛查并坚持低Phe饮食至成人,另1例也是新生儿筛查发现,但直到20个月才开始治疗,说明虽然治疗开始晚,但坚持治疗,控制好血Phe水平,可以具有健康人的智力水平,有能力很好地管理自己和胎儿的营养状况。Koch等[10]也阐述了PKU母亲智力与胎儿发育的关系,如果母亲的智商<85,就需要有经验的营养师和护士参与指导饮食,监测血Phe浓度。
Lee等[11]分析了英国PKU的随访数据,在孕前即控制饮食的PKU母亲与孕后才开始控制饮食的PKU母亲相比,其婴儿出生时有较高的体质量、较大的头围,较少患先天性心脏病,智力正常,发育良好。
PKU母亲所产的先天缺陷患儿虽然是PAH基因突变携带者,但往往不是PKU患儿。由此可见,为了保证下一代健康出生,必须全程严格控制PKU母亲的饮食及血Phe浓度。
为避免母源性PKU的发生,对准备怀孕的女性PKU患者,至少从孕前3个月开始,严格控制血Phe浓度,直至整个孕期结束,将血Phe浓度控制在120~360 μmol/L。为了有效地控制PKU孕母的血Phe浓度,国内外形成了一些共识。
首先,婚育期女性PKU患者如准备怀孕或已经怀孕,应及时寻求有经验的遗传学医师及营养师的帮助,获得合理的个体化饮食指导及营养代谢监测。
为保证母婴胎儿健康,每月至少复查1次,测量体质量,计算体质量指数(BMI);每周1~2次检测血Phe浓度;每周1次(至少每月1次)进行血氨基酸分析,每月1次(至少每孕期1次)进行血清前蛋白测定。每个孕期均要行清蛋白、总蛋白、血常规、铁蛋白、25(OH)维生素D的检测,如条件许可,还可进行血液维生素B12、维生素B6、维生素A、叶酸、锌、铜、硒及必需脂肪酸测定,以全面掌控PKU孕母的血Phe浓度和全面营养状况,并根据监测情况实时对饮食方案及相关治疗进行调整,见表1[12]。
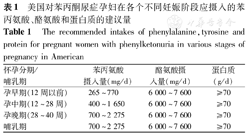
美国对苯丙酮尿症孕妇在各个不同妊娠阶段应摄入的苯丙氨酸、酪氨酸和蛋白质的建议量
The recommended intakes of phenylalanine,tyrosine and protein for pregnant women with phenylketonuria in various stages of pregnancy in American
美国对苯丙酮尿症孕妇在各个不同妊娠阶段应摄入的苯丙氨酸、酪氨酸和蛋白质的建议量
The recommended intakes of phenylalanine,tyrosine and protein for pregnant women with phenylketonuria in various stages of pregnancy in American
| 怀孕分期/哺乳期 | 苯丙氨酸摄入量(mg/d) | 酪氨酸摄入量(mg/d) | 蛋白质(g/d) |
|---|---|---|---|
| 孕早期(12周以前) | 265~770 | 6 000~7 600 | ≥70 |
| 孕中期(12~28周) | 400~1 650 | 6 000~7 600 | ≥70 |
| 孕晚期(28~40周) | 700~2 275 | 6 000~7 600 | ≥70 |
| 哺乳期 | 700~2 275 | 6 000~7 600 | ≥70 |
为保证母婴营养支持,需添加叶酸、维生素B12和蛋白质,使胎儿血浆总同型半胱氨酸、叶酸、基础长链脂肪酸维持在正常水平[13],蛋白供给充分,降低心血管疾病的风险,改善骨密度,减少宫内发育迟缓[14]。
许多PKU孕妇饮食治疗极其困难,与普通孕妇一样,PKU孕妇有呕吐等孕期不适,不能忍受甚至拒绝低Phe配方奶粉等特殊饮食。为此,需寻找其他替代疗法。最近,国外将用于治疗BH4缺乏症的药物沙丙蝶呤(sapropterin dihydrochloride,6R-BH4)用于BH4反应型的HPA孕母,取得了一些效果。欧美、日本已将6R-BH4作为BH4反应型PKU孕妇的饮食治疗补充剂[7]。
6R-BH4是BH4的生物活性合成形式。BH4反应型HPA患者约占全部PKU患者的50%或以上,在服用6R-BH4后,PAH的活性得以改善。BH4是一氧化氮合成酶的辅酶,能够调节胎盘至胎儿的血流量,增强母体向胎儿输送营养素和氧的能力。营养过剩和营养不足均可影响一氧化氮的合成。在BH4反应型PKU患者中,BH4治疗能够增加天然蛋白的摄入容忍量,降低血Phe浓度水平,激活胎儿的PAH活性。
Koch等[15,16]首次将6R-BH4用于1例计划怀孕的PKU孕妇,当时使用的剂量未超过100 mg/d,其子4岁时的发育商为132分。Koch[16]建议孕早期6R-BH4的服用剂量为100 mg/d,孕中期可提高至200 mg/d,妊娠后期剂量可为300 mg/d。2009年Moseley等[9]又报道了2例计划怀孕的PKU孕妇,均产下健康新生儿。2例孕妇在孕中期和孕晚期血Phe浓度均显著下降,甚至低于正常值,可见随着孕期的进展,天然蛋白的摄入也需不断增加,意味着Phe的摄入也需不断增加,以保证胎儿的正常发育。BH4的使用增加了患者对天然蛋白的摄入容忍量,在整个怀孕期间使用6R-BH4,剂量达到600 mg/d,未出现任何不良反应。
西班牙的Aldámiz-Echevarría等[17]报道了1例BH4反应型PKU孕妇,服用6R-BH4产下健康婴儿。患者怀孕前已连续使用6R-BH4[10 mg/(kg·d),即500 mg/d]4年,在最易出现畸形的孕早期(前12周)6R-BH4的最大剂量为500 mg/d;妊娠第1个月血Phe浓度能控制在理想水平;在孕中期至孕后期,胎儿PAH被激活,血Phe浓度得以降低;至孕中期的第20周,6R-BH4的使用剂量减至250 mg/d。至孕后期的第28周,再减量至100 mg/d,直至整个孕期。在使用期间未发现任何不良反应。患者于妊娠41周时分娩,新生儿健康,此后体格及神经系统均发育良好。
2014年美国总结了一项关于PKU孕妇的研究(PKU MOMS)[7],21例PKU孕妇入选,其中5例在怀孕前使用了6R-BH4治疗,16例怀孕期间使用了6R-BH4治疗,除4例自然流产的孕妇外,孕期使用6R-BH4的14例孕妇血Phe浓度为(204.7±126.6) μmol/L,而怀孕前开始使用BH4的3例孕妇孕期血Phe浓度为(267.4±300.7) μmol/L,怀孕期间使用BH4的孕妇较怀孕前使用BH4的孕妇血Phe浓度降低23%。BH4属于孕妇C类药,虽然BH4可联合低Phe饮食治疗BH4反应型PKU患者,但是用于PKU孕妇的循证医学证据有限。Grange等[7]认为,由于BH4导致的不良反应事件可能包括1例早产、1例自然流产和1例35+4周早产伴婴儿食欲过盛。同年,欧洲也报道了8例PKU孕妇使用BH4的研究结果,剂量为4~20 mg/(kg·d)[18]。8例患者在怀孕前血Phe浓度均高于各国推荐的理想控制浓度,除第8例外,其余7例在整个孕期均控制在各国推荐的浓度范围之内。8例孕妇均进行了基因检测,其中7例至少有1个基因突变与BH4反应型相关,第8例未携带与BH4反应有关的基因。7例对BH4治疗有反应的孕妇使用BH4以后血Phe浓度均得到了有效的控制,并增加了Phe摄入的容忍量,从400 mg/d增加到800 mg/d,7例均产下了正常出生的新生儿,并未出现BH4的不良反应。第8例在妊娠前未进行饮食控制,未进行BH4负荷试验,发现怀孕时已8周,BH4只是作为补救的治疗,妊娠16周时超声波检查发现羊水减少,于33周早产,伴Potter综合征,出生后1 h夭折。胎儿早产及Potter综合征的原因考虑与母亲孕早期血Phe控制不佳有关。
关于BH4反应型HPA,我国进行过相关的临床与基因研究,发现部分因PAH缺乏引起的中轻度PKU患者为BH4反应型,给予BH4治疗可部分或全部替代低Phe饮食治疗,提高患者生活质量[19,20]。PKU为罕见病,目前尚缺乏大量BH4用于PKU孕妇的数据,暂不提倡BH4作为一线药物使用,但对于饮食治疗困难的BH4反应型PKU孕妇,BH4可以作为妊娠前和妊娠期的补充治疗。为此,对准备生育的PKU患者需首先应进行BH4负荷试验和基因检查,判断其是否为BH4反应型,从而考虑是否使用BH4来降低其血Phe浓度。
对于计划生育子女的PKU患者及其家庭,美国妇产科协会提出了如下一些建议[4]:
(1)鼓励终身进行饮食控制及治疗,改善生活质量。(2)婚育年龄的女性PKU应接受遗传知识方面的教育,内容包括结婚对象的选择、家庭婚育计划及关于母源性PKU的相关问题。(3)准备怀孕的女性患者孕前至少3个月起控制血Phe浓度低于360 μmol/L,自孕前直至整个妊娠期间血Phe水平需维持在120~360 μmol/L。(4)评估妊娠导致早期发生骨量减少的风险。(5)分娩后要兼顾好治疗和营养2个方面。(6)怀孕PKU女性应在富有经验的PKU治疗中心进行监控与营养干预。(7)如果婴儿不是PKU患儿,其肝脏内PAH活性正常,可以顺利代谢Phe,故母乳喂养是安全的。(8)尽管研究资料有限,对于BH4反应型的PKU孕妇,可以选择服用6R-BH4作为饮食治疗的补充。
既往30余年新生儿筛查研究经验证实PKU是我国发病率最高的遗传代谢病之一,随着新生儿筛查的普及,患者的生存质量显著提高,成年PKU患者的管理及生育问题将成为重要的课题[1]。PAH基因定位于染色体12q22-24.1,全长约90 kb,含13个外显子,编码451个氨基酸,至今国内外已报道近800种PAH基因突变,具有高度的异质性,存在着显著的地区和人种差异及个体差异,我国各地进行了广泛的临床和遗传学研究[21,22,23]。
我国人群PAH突变基因携带率约为1/60以上,对于有生育要求的PKU妇女,最好能选择与不携带PAH基因突变的对象结婚,如果想生育健康的孩子,至少孕前3~6个月,将血Phe浓度控制在120~360 μmol/L,并补充叶酸等营养素。为了将整个孕期血Phe浓度控制在理想范围,严格的低Phe饮食控制非常重要。对BH4反应型PKU孕妇,在饮食治疗困难或严格的低Phe饮食不能控制血Phe浓度的情况下,可以考虑使用BH4。
感谢北京大学第一医院儿科杨艳玲教授、中日友好医院临床医学研究所赵世萍研究员对本工作的指导

























