
急性白血病是我国最常见的儿童恶性肿瘤,其发病机制包括原癌基因的转化、抑癌基因畸变、细胞凋亡受抑等。在致癌因素的作用下,染色体通过点突变、缺失、重排或基因扩增等方式,导致原癌基因及抑癌基因的结构变异,产生新的融合基因。这些基因中有些是调控细胞增殖、分化和衰老、死亡的转录因子,当基因发生变异,直接影响了下游信号传递途径,导致细胞增殖增强、凋亡和/或分化障碍等,从而导致白血病的发生。近年来,基因测序技术的发展使越来越多的白血病预后基因展现在公众视野,并应用于临床治疗及评估预后、复发等中。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
近10年来,由于基因测序技术的发展,特别是二代测序、全基因组测序、全外显子测序、RNA测序,越来越多的白血病预后基因得以发现并应用于临床,特别是对一些复发、难治白血病的全基因组测序,发现了更多新的有价值的基因变异,除常规的基因分析如急性淋巴细胞白血病(ALL)的TEL-AML1、E2A-PBX1、MLL-AF4、BCR-ABL1、PDEGFRb重排、C-myc重排等;急性髓系白血病(AML)常见的融合基因有BCR-ABL、AML1-ETO(RUNX1/T1RUNX1)、PML-RARa(M3)、CBFβ-MYH11、inv(16)或t(16;16);新的基因变异和对预后的影响不断发现和被临床采纳,如酪氨酸激酶(JAK)通路相关基因、JAK基因突异、转录因子基因异常、NPM1突变、WT1突变、FLT3-ITD等,现对此进行综述。
即CRLF2基因异常,包括CRLF2/IGH易位(CRLF2过表达),CRLF2-P2RY8易位(CRLF2过表达),CRLF2点突变(F232C)。
CRLF2位于染色体Xp22.3/Yp11.3假常染色体区1(PAR1)上,编码胸腺基质淋巴细胞生成素受体(thymic stromal lymphopoetin receptor,TSLPR),在配体TSLP存在情况下,TSLPR和IL-7Ra形成异源二聚化的受体复合物并活化,启动下游的JAK-STAT信号通路,参与调控细胞的发育和增殖,导致细胞增殖过表达和活化突变,近年来有文献报道,CRLF2的基因高表达与B系淋巴细胞白血病的复发及不良预后相关[1]。
CRLF2基因定位距离端粒的长度仅为1 Mb,而免疫球蛋白重链位点(IGH@)位于染色体14q的端粒,因此距离相近的两者有遗传学易位的潜在可能性。其改变包括Xp/Yp并列到14q32(IGH@),从而产生重排[2]。Russell等[3]报道在B系淋巴细胞白血病患者中大约有5%的患者在Xp22.3或Xp11.3IGH@易位到CRLF2或CRLF2上游缺失,导致CRLF2过表达,引起B系淋巴细胞白血病的复发。
约50%伴CRLF2易位的Ph样ALL患者中可检测到JAK基因突变,B淋巴细胞急淋白血病(B-ALL)中的JAK基因突变也主要见于该组患者,以JAK2R683突变最多见,提示CRLF2易位和JAK基因在活化JAK-STAT通路中起着协同作用,可能多个因素的并存是必须的。
约7.4%的Ph样ALL,尤以JAK2易位多见;易位通常保留了JAK基因的激酶结构域,并有融合蛋白形成;JAK的伙伴基因多样;这些易位通过激活JAK-STAT信号通路,导致细胞持续增殖。
常有其他JAK-STAT通路基因(TSLP、CRLF、IL-7R、TYK2、SH2B3等)的突变,偶有FLT3等其他激酶基因突变(说明:仅有CRLF2易位或表达不足以导致Ph样ALL,还需要其他因素协同作用)。
可仅携带IL-7Ra、FLT3、JAK1、JAK3突变和SH2B3缺失等JAK-STAT通路基因的异常,也能导致该通路的异常激活。
见于约3.9%的Ph样ALL,其伙伴基因主要是IGH或IGK;EPOR与其配体结合,活化并传递信号给JAK激酶,启动JAK-STAT信号通路。
60%的B-ALL患者可检测到B系发育相关转录因子(transcription factors,TF)基因的异常,常见的有IZKF1、PAX5、PRPS1、TCF3等。
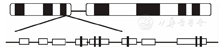
IKZF1基因位于7p12.2,含8个外显子,大小约100 320 bp,编码519个氨基酸(图1)。其中外显子1不参与转录,可能与启动子一起参与调节其他基因的转录,而外显子2、3和7的功能尚未完全了解。其余的外显子对于行使正常的IKAROS功能至关重要,4~6号外显子编码N末端4个结合DNA所必需的锌指结构,8号外显子编码C末端2个用于IKAROS二聚化或与其家族其他成员结合的锌指结构[8,9]。IKZF1基因编码的IKAROS蛋白主要位于细胞核,其在淋巴细胞分化过程中起到2个重要的调节作用:(1)为造血干细胞-多能造血祖细胞提供淋巴细胞分化潜能;(2)在前体T和B淋巴细胞的第二抗原受体链重组和T、B淋巴细胞选择的分化增殖阶段,使其处于静止状态。一些关于IKAROS功能的研究证实,IKAROS不仅参与淋巴细胞的正常分化,而且参与IKZF1突变导致的高危白血病进展。
已有许多研究发现,在一些IKZF1突变所致的特定SNPs可诱发不同类型的白血病:T淋巴细胞ALL(T-ALL)(rs11978267)[10],婴幼儿和儿童AML(rs11978267纯合子)[11,12],成人前体B淋巴细胞ALL (rs4132601)[13]及最常见的儿童前体B淋巴细胞ALL (rs4132601)[14,15]。IKZF1还存在一些有意义的其他异常改变,如IKZF1启动子区的过度甲基化,其可见于大多数大肠腺癌,易位:t(3;7)(q27;p12)在弥散性大B淋巴细胞淋巴瘤(DLBCL)中反复出现。
PAX(paired box)基因因含有一共同编码128个氨基酸的配对结构域而得名,是一组贯穿于整个进化过程中高度保守的发育调控基因,在哺动物发育过程中起重要作用。PAX5是其中一员,定位于9q13,编码PAX-5蛋白,又称B淋巴细胞特异性活化蛋白(B cell specific activation,BASP),在B淋巴细胞的发育、激活及分化中起重要作用,是B淋巴细胞发育和活化所必需的转录因子。其表达始于B淋巴细胞发育的早期,贯穿于从原B淋巴细胞到浆细胞前的整个B淋巴细胞发育阶段,随着B淋巴细胞逐渐向浆细胞分化,其表达下调。近年来,PAX5基因的点突变、缺失和重排已经在人类数种癌症中被报道。在Ph样ALL中主要与激酶基因易位形成融合基因,如PAX5-JAK2有阻滞细胞分化和促进增殖的双重作用。
PRPS1即磷酸核糖焦磷酸合成酶-1,是细胞内嘌呤代谢合成途径的限速酶和变构调节酶,由3个同源二聚体形成螺旋桨样的六聚体;PRPS1基因突变与ALL的复发相关,有研究报道PRPS1基因突变通过减少从头嘌呤生物合成的反馈抑制作用诱导ALL药物治疗的耐药性[16]。
IL-7R基因突变见于7%B-ALL及T-ALL;IL-7R突变导致IL-7及JAK-STAT信号途径激活。
表现调节基因突变,在早期前体T-ALL发生的频率很高,也可见于滤泡淋巴瘤(FL),在这2种疾病,EZH2突变的位点不同,在FL多位Y641突变,提示可用表观基因调节剂治疗。
Notch信号与肿瘤的关系最早发现于T-ALL,t(7;9)(q34;q34.3)染色体易位使9号染色体上NOTCH1基因的胞内段编码区与7号染色体上T淋巴细胞受体(T cell receptor,TCR)β基因的增强子和启动子区融合,导致Notch信号组成型激活,引起了肿瘤的发生。最近有学者报道,约50%的T-ALL患者存在NOTCH1激活的突变,说明NOTCH1的异常激活是T-ALL的重要发病机制[17]。
NOTCH1激活性突变见于约40%的T-ALL;FBXW7及NUMB为NOTCH的调节基因,如果发生功能丢失性突变也可导致NOTCH1激活;最近已经开发了NOTCH1的小分子抑制剂或抗体,可能对这类基因突变患者有效。
ZNF384基因被报道与B-ALL相关。ZNF384基因即锌指蛋白384,编码一种转录基因,调节细胞外基质基因的启动子,是通过与TET家族基因的融合参与到ALL的调节中,如EWSR1基因[EWSR1基因,t(12;22)],TATA盒结合蛋白相关因子[TAF15,t(12;17)],转录因子3[TCF3或E2A,t(12;19)][18]。
AML是一种临床和生物学的异质性疾病。正常核型AML(CN-AML)是最大的一个亚群,代表约45%的成人患者,在过去的几年中,新兴的数据表明,体细胞突变与治疗结果相关,作为分子分层指导风险评估和治疗的基础。
WT1在AML的发生率约为10%,并且是AML的潜在标志物。WT1基因位于11p13染色体上,编码一个锌指转录因子,监管正常和恶性的造血作用。积累数据显示,WT1似乎是一个致癌基因而不是抑癌基因[23]。WT1基因在大多数AML中有过表达,但有关它能否成为AML-微小残留病(MRD)检测中的光谱分子标志物仍有争议。WT1区分低水平MRD与正常背景的能力有限,并且外周血似乎较骨髓标本更适合WT1表达检测,因为外周血背景值更低;WT1-MRD检测可以提供一定程度的独立的预后意义,但由于其相对低的敏感性和缺乏白血病的特异性,这一指标不可能成为临床广泛应用的MRD检测途径。另外,将WT1作为移植后MRD检测指标在早期存在着一些争议,但随着近年技术的进步以及人们对WT1基因认识的加深,WT1表达水平与移植后患者疾病进展相关性已越来越受到重视[24]。
NPM1又称B23、N038,是位于核仁颗粒区的一种在各种类型的细胞中广泛表达的多功能蛋白质。目前的多项研究发现,NPM1突变在AML中,尤其是在正常核型AML(nk-AML)中检出率最高的一种分子遗传学异常。NPM1突变是AML一类特殊的亚群,具有相对独特的临床特点,且是AML独立的预后良好的指标[25]。
NPM1基因突变一般导致NPM1在AML中高表达;MRD检测能预测复发风险:治疗后持续的高表达或分子缓解后出现表达增高的现象都预测复发风险的增加。
EWSR1融合各种伴侣基因以促进尤文肉瘤家族的发展。与此相反,EWSR1嵌合基因与白血病的关系鲜见报道。研究发现一种新的EWSR1相关嵌合融合基因在AML中隐藏46,XY,t (11; 22) (p13; q12)核型异常。该类患者很难强化治疗,甚至是造血干细胞移植。总RNA的双末端测序鉴定了一种新的嵌合基因的EWSR1/ELF5,该基因属于E26转换特定成员(ETS)转换基因家族。将EWSR1/ELF5转导的NIH3T3细胞系统地注入NSG-SCID小鼠中,诱导肿瘤发展。这些结果揭示了EWSR1/ELF5的致癌潜能[26]。
FLT3属于Ⅲ型受体酪氨酸激酶家族成员,近年来,许多大样本研究已经证实FLT3的激活突变在AML的发生及疾病的进展中导致疾病恶化。
FLT3的激活突变主要有2种:ITD在AML和MDS患者中的发生率分别为15%~35%和5%~10%,在ALL中的发生率<1%,且主要见于双表型的ALL病例;活化环中的点突变(point mutation in the activation loop,TKD),在AML、MDS和ALL患者中的发生率分别为5%~10%、2%~5%和1%~3%。FLT3的这2种激活突变均能引起FLT3发生自动磷酸化进而导致FLT3发生配体非依赖性的组成性激活,进一步激活其下游异常的信号转导,从而起到促进增殖和抑制凋亡的作用,使得具有此突变表型的白血病患者临床预后较差。由于FLT3-TKD的发生率较低,且与其相关的临床特征尚不明确,但FLT3-ITD的临床特征却很明确,具有FLT3-ITD激活突变的AML患者通常具有外周血白细胞计数高、临床预后较差、易复发等独特的临床特征,并且由于FLT3激活突变的检测方法简单易行,故有越来越多的研究者致力于将FLT3发展成为AML患者常规的检测手段用来指导AML患者的治疗和预后的判断以及作为MRD的检测手段,并将其作为白血病患者化疗药物的又一新的靶点(目前已经有针对FLT3-ITD突变进行治疗的药物)。
白血病是一种造血干细胞异常克隆的增殖性疾病,其发病多与基因突变及融合基因相关。检测融合基因及基因突变的方法主要包括荧光原位杂交、免疫印迹、聚合酶链反应、流式细胞术、基因芯片及全基因组测序等方法。这些检测方法的应用大大提高了基因检测的阳性率,对进一步明确急性白血病的分型、危险度、治疗及预后起重要意义。

























