
巨噬细胞活化综合征(macrophage activation syndrome,MAS)是一种严重的具有潜在生命威胁的、多继发于幼年特发性关节炎全身型(systemic onset juvenile idiopathic arthritis,sJIA)失控的自身炎症性反应综合征。其临床表现包括持续发热、肝、脾淋巴结大、血细胞减少、高铁蛋白血症、低纤维蛋白原血症、高三酰甘油血症、凝血功能障碍以及骨髓中可见吞噬血细胞。现总结继发于sJIA的MAS的临床特征,并对其诊断和治疗上的新进展进行综述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
巨噬细胞活化综合征(MAS)是一种严重的具有潜在生命威胁、多继发于幼年特发性关节炎全身型(sJIA)失控的自身炎症性反应综合征。1985年Hadchouel第1次描述继发sJIA的以出血、肝炎和神经系统异常表现的综合征,直到1993年发现了巨噬细胞活化的证据,并注意到MAS与噬血细胞综合征(HLH)有着极其相似的临床表现,第1次提出了MAS的概念并归纳到继发性HLH范畴。现总结继发于sJIA的MAS的临床特征,并对其诊断和治疗上的新进展进行综述。
继发于sJIA的MAS发病率并不十分清楚,有7%~13%的sJIA患儿发生MAS[1],30%~40%的sJIA患儿合并亚临床型MAS或轻型MAS,亚临床MAS概念的提出有助于对这种严重并发症的早期识别和早期治疗。一项来自全世界33个国家、99名儿童风湿科医师参与的多中心研究对392例继发sJIA的MAS的临床和流行病学特征进行分析[2],显示MAS似乎更多发生于女童,与男童比例为6︰4;sJIA起病年龄中位数为5.3岁;并发MAS的时间在起病3.5个月;近22%的MAS与sJIA同时被诊断,提示MAS可作为sJIA的早期表现甚至首发症状。
免疫反应失控导致细胞毒性T淋巴细胞和巨噬细胞过度激活和增殖是MAS的典型病理学特征[3],激活的免疫细胞产生大量炎性因子,形成高细胞因子血症。MAS的临床特征和家族性噬血细胞综合征(FHLH)相似,FHLH是一种常染色体隐性遗传性疾病,大约30%的FHLH存在穿孔素基因突变,导致被病毒感染的靶细胞、肿瘤细胞及活化的淋巴细胞自身凋亡途径受阻。10%的FHLH存在与穿孔素释放相关基因MUNC13-4的突变[4]。近期有报道,穿孔素颗粒细胞内转运基因(STX11)及突触融合蛋白2(STXBP2)基因的突变与FHLH相关[5]。这类患者中的细胞毒性细胞能够产生足量的穿孔素,但是由于释放过程以及与靶细胞融合受阻,其杀伤靶细胞的功能显著下降,这样会延长活化的CD8+T淋巴细胞、自然杀伤(NK)细胞和感染的靶细胞之间相互作用时间,最终导致促炎细胞因子风暴反应。
淋巴绒毛样脑膜炎病毒感染动物能够诱导HLH模型,出现典型的发热、全血细胞减少、铁蛋白增高及组织噬血现象,而这些HLH特征可以通过注射CD8抗体或γ-干扰素(IFN-γ)中和抗体而阻断。HLH及MAS患者肝脏组织活检也证实大量分泌IFN-γ的CD8+T淋巴细胞及分泌肿瘤坏死因子-α(TNF-α)、白细胞介素(IL)-6的巨噬细胞浸润。MAS时均伴可溶性白细胞介素2受体(sIL-2Rα)增高,用环孢素治疗MAS有效,而环孢素直接抑制T淋巴细胞,对巨噬细胞作用不大,这些均佐证了分泌IFN-γ的CD8+T淋巴细胞在HLH发病机制中起重要作用。野生型小鼠在Toll样受体-9(TLR-9)的重复刺激并联合IL-10阻滞剂的共同作用下可以出现典型的MAS特征,表明TLR/IL-1β通路高度活化和IL-10功能下降是发生MAS的重要免疫学基础。
MAS能够快速进展为多器官衰竭,病死率高达8%,因此,从活动性sJIA中早期识别MAS就显得格外重要。但是MAS的临床特征与sJIA的疾病活动或严重的全身性感染状态有很多重叠,MAS临床表现具有很强的异质性,尚无一个单一的病理或实验室指标确诊,仅有60%的MAS组织病理学结果提示噬血现象,而且多出现在疾病的中后期,这样,在临床工作中就迫切需要一些诊断标准将MAS从活动性sJIA或其他疾病状态中区分出来。
MAS起病迅速并很快进展为多器官障碍,对于任何已经诊断或疑似诊断sJIA的患者都必须全程监测相应的MAS特征和实验室检查。发热是MAS和sJIA共同的临床特征,但sJIA为弛张热,而MAS多表现为持续发热不退,当由典型的sJIA弛张热转变成持续的、不能缓解的发热时,应警惕发生MAS。肝、脾淋巴结大在发生MAS时会进一步加重。MAS中枢神经系统功能障碍更为常见,表现为乏力、易激惹、定向障碍、头痛、抽搐甚至昏迷。20%的患者出现皮肤黏膜瘀点、瘀斑甚至黏膜出血。MAS容易引起肺间质性病变而导致呼吸衰竭,心力衰竭和肾功能不全较为少见。重要脏器受累提示预后不良甚至死亡,多器官损害、中枢神经系统受累、血红蛋白低于7.9 g/L、年龄>11.5岁是MAS预后不良的独立危险因素[6]。MAS的临床特征具有很强的异质性,部分患者发生MAS时并不发热,而是以重症肝炎为突出表现[7],这类患者在给予环孢素治疗后肝功能迅速好转,因此,对于sJIA患者出现肝功能急剧恶化,即使无发热,也应积极检测MAS相关指标并给予治疗。
MAS典型的实验室特征主要有全血细胞减少,包括白细胞、血小板和血红蛋白的降低,转氨酶增高和胆红素异常,轻度低清蛋白血症,凝血功能异常包括凝血酶原时间和部分凝血时间延长、低纤维蛋白原血症、D-二聚体增高。血生化提示三酰甘油和乳酸脱氢酶增高。血清铁蛋白异常增高(5 000~数万μg/L)是MAS的重要标记之一,动态检测SF的变化对于监测疾病活动、评价治疗效果和提示预后尤为重要[8]。sJIA发生MAS时关节炎、浆膜炎的症状和体征可以减轻甚至消失,原因不明。MAS时红细胞沉降率(ESR)降低或接近正常,这与低纤维蛋白血症有关。血清铁蛋白和ESR的比率(SF/ESR)>80也可作为有效区分MAS和sJIA的指标,敏感性和特异性高达100%[9]。低补体血症在MAS中也有报道,其发病机制可能与血浆中的纤溶酶激活有关[10],而其他炎症指标,如CRP则持续增高。以上实验室指标的变化多在MAS典型的临床症状之前出现,而大多数MAS或亚临床MAS的早期诊断都依赖于实验室检查。在MAS发生前后变化超过50%的实验室指标依次排序为:血小板、转氨酶、血清铁蛋白、乳酸脱氢酶、三酰甘油和D-二聚体。2013年美国风湿病协会(ACR)对幼年特发性关节炎治疗建议(2011)的更新中提出,同时具备发热、血小板计数直线降低或其他实验室指标任意2项就可以归类为"具有MAS特征的sJIA" ,可见血小板降低对于MAS的早期预警尤为重要。
sJIA是以固有免疫失调导致的一类自身炎症性疾病,细胞因子(IL-1、IL-6、IL-18)的增高是导致sJIA全身症状出现的病理生理基础。虽然针对IL-6的单克隆抗体(TCZ)可以缓解sJIA的全身症状,但是仍然有患者在TCZ治疗同时发生MAS,这表明sJIA是一类高度异质性疾病并存在一些临床亚型。Shimizu等[11]根据IL-18和IL-6的比值将sJIA分为2种临床亚型:IL-18/IL-6<1 000的sJIA容易有更为严重的关节炎,IL-18/IL-6>1 000则更倾向发生MAS。sJIA发生MAS的IL-18水平异常增高,而IL-6水平在活动性sJIA和MAS之间并无明显的变化,认为IL-18>47 750 ng/L是发生MAS的截点。与EBV相关性HLH及川崎病、红斑狼疮并发的MAS相比,仅有sJIA并发MAS出现了IL-18异常增高,因此,IL-18可作为预测sJIA发生MAS的重要血清学指标。
可溶性CD25(sCD25)和可溶性CD163(sCD163)直接反映T淋巴细胞和巨噬细胞的激活和增殖,在亚临床MAS时其血清水平显著增加,与增高的SF、正常血小板、低血红蛋白及NK细胞功能下降等具有明显相关性,这些变化甚至出现在细胞因子风暴之前,因此,认为sCD25和sCD163可以作早期指标识别亚临床MAS。卵泡抑素相关蛋白-1(FSTIL-1)在sJIA/MAS时明显增加,血清和尿中的β2微球蛋白水平也显著增加。活动性sJIA和MAS的临床和实验室指标的区别见表1[12]。
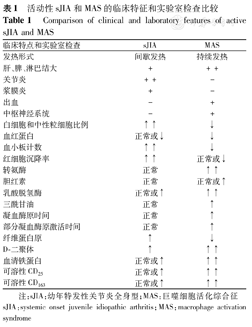
活动性sJIA和MAS的临床特征和实验室检查比较
Comparison of clinical and laboratory features of active sJIA and MAS
活动性sJIA和MAS的临床特征和实验室检查比较
Comparison of clinical and laboratory features of active sJIA and MAS
| 临床特点和实验室检查 | sJIA | MAS |
|---|---|---|
| 发热形式 | 间歇发热 | 持续发热 |
| 肝、脾、淋巴结大 | + | ++ |
| 关节炎 | ++ | - |
| 浆膜炎 | + | - |
| 出血 | - | + |
| 中枢神经系统 | - | + |
| 白细胞和中性粒细胞比例 | ↑↑ | ↓ |
| 血红蛋白 | 正常或↓ | ↓ |
| 血小板计数 | ↑↑ | ↓ |
| 红细胞沉降率 | ↑↑ | 正常或↓ |
| 转氨酶 | 正常 | ↑↑ |
| 胆红素 | 正常 | 正常或↑ |
| 乳酸脱氢酶 | 正常或↑ | ↑↑ |
| 三酰甘油 | 正常 | ↑ |
| 凝血酶原时间 | 正常 | ↑ |
| 部分凝血酶原激活时间 | 正常 | ↑ |
| 纤维蛋白原 | ↑ | ↓ |
| D-二聚体 | ↑ | ↑↑ |
| 血清铁蛋白 | 正常或↑ | ↑↑ |
| 可溶性CD25 | 正常或↑ | ↑↑ |
| 可溶性CD163 | 正常或↑ | ↑↑ |
注:sJIA:幼年特发性关节炎全身型;MAS:巨噬细胞活化综合征
sJIA:systemic onset juvenile idiopathic arthritis;MAS:macrophage activation syndrome
在早期甚至现在仍然有部分医师参照HLH-2004诊断标准[13],标准中8条有5条符合即可诊断。但是该标准有很多局限性,sJIA是一种自身炎症性疾病,血小板、白细胞、血红蛋白、纤维蛋白原等指标在发生MAS时表现为相对降低,不能满足标准中这些指标数值的绝对降低。MAS急性期血清铁蛋白峰值高达5 000 μg/L以上,而诊断标准中SF值仅为500 μg/L,不能有效区分MAS和sJIA疾病活动。NK细胞活性、sCD25等指标多数医院不作为常规检查。目前应用更为广泛的是2005年Ravelli等[14]提出的sJIA并发MAS初步诊断指南:(1)实验室指标包括血小板减少(≤262×109/L)、天冬氨酸氨基转移酶升高(>59 U/L)、白细胞计数下降(≤4.0×109/L)、低纤维蛋白原血症(≤2.5 g/L);(2)临床指标包括中枢神经系统功能障碍(激惹、头痛、昏睡、定向力障碍、抽搐、昏迷)、出血(紫癜、黏膜出血)、肝大(肋缘下≥3 cm);(3)组织病理学标准(骨髓涂片证实有巨噬细胞噬血现象)。符合≥2条实验室指标或至少1条实验室指标和1条临床指标,可考虑MAS,骨髓检查发现嗜血现象仅用于疑似病例的诊断。研究显示该诊断标准的敏感性和特异性高达86%。
儿童风湿病国际研究组织(PRINTO)最近完成了一项sJIA并发MAS的分类标准,该研究首先通过Delphi调查技术,由全世界47个国家的232名儿童风湿病专家确认了9项有可能作为MAS分类标准的临床和实验室指标,然后通过儿童风湿病专家和血液肿瘤专家对纳入网站的1 111例患者的数据进行回顾性队列研究,提出一系列候选sJIA/MAS分类标准并进行统计学分析,于2014年3月份在意大利热那亚召开了由28名专家参加的国际MAS分类标准统一大会,通过投票方式最终确立了sJIA/MAS的分类标准[15]:伴发热的确诊或疑似sJIA患者如满足以下条件考虑并发MAS:血清铁蛋白>684 μg/L,同时具备下列4项指标中任意2项:(1)血小板减少(≤181×109/L);(2)天冬氨酸氨基转移酶升高(>48 U/L); (3)三酰甘油升高(>1 560 mg/L); (4)纤维蛋白原降低(≤3 600 mg/L)。新的分类标准将发热和铁蛋白视为必须条件,增加了三酰甘油指标,但却降低了血小板的预警值,由于这一新的分类标准源自专家的意见,且忽视了生物制剂的潜在影响,其临床有效性仍有待验证。
目前MAS多采用高剂量激素联合T淋巴细胞免疫抑制剂治疗。由于MAS起病急骤且进展迅猛,需要早期应用起效迅速和作用强大的免疫抑制剂控制炎性反应,低剂量激素治疗MAS无效。大剂量激素冲击治疗迅速控制炎性反应,缓解病情,是治疗MAS的首选药物。环孢素能明显抑制T淋巴细胞活性,对MAS患者起"拯救性"作用,初期采取静脉给药方式,待病情稳定后改为口服,尚不清楚MAS临床症状改善后环孢素需要维持多久,突然停药会引起MAS复发,用药期间应监测其血药浓度。依托泊苷(VP16)是HLH-2004化疗方案中最为重要的药物,但对于继发风湿病的MAS其作用并不明确,且绝大多数MAS在激素联合环孢素治疗后都能缓解,无需应用VP16治疗。尽管大剂量丙种球蛋白冲击治疗MAS应用较为广泛,但其疗效并不确定。
生物制剂治疗sJIA效果显著,但是能否使sJIA/MAS获益仍有争议。有报道IL-1受体拮抗剂(Anakinra)可显著改善对传统治疗失败的sJIA/MAS的临床症状,但是在另一项较大规模的应用IL-1受体拮抗剂治疗sJIA的研究中,似乎是生物制剂诱导了其中5例患者发生了MAS,多数病例在增加IL-1受体拮抗剂剂量后临床症状缓解[16,17]。尽管2013北美治疗sJIA建议更新中推荐应用IL-1受体拮抗剂初始治疗具有MAS特征的sJIA,但将其作为难治性MAS治疗方案仍需要更多的临床证据,IL-6受体拮抗剂是国内唯一的针对IL-6的细胞因子拮抗剂,能够有效控制sJIA系统性炎症,迅速退热,但是其改善MAS临床特征的作用有限[18,19,20]。MAS的本质是多种细胞因子风暴,其炎症程度远远超出sJIA基础疾病,从理论上讲通过单纯阻断个别细胞因子并不一定能够阻断过度失控的炎性反应。因此,细胞因子拮抗剂治疗sJIA/MAS可作为传统治疗的有益补充,今后仍需要大量临床研究才能确定其在sJIA/MAS中的治疗价值。

























