
探讨伴低血糖脑病的果糖-1,6-二磷酸酶缺乏症患儿的临床特点、基因突变特点、治疗及预后。
总结北京大学第一医院儿科收治的1例伴低血糖脑病的果糖-1,6-二磷酸酶缺乏症患儿的临床表现、基因突变及其他辅助检查特点、治疗及预后。
患儿,男,2岁5个月时因不明原因脑病、肝病就诊。患儿发病前发育正常。1岁3个月时进食甜瓜后呕吐、腹泻、腹痛。1岁3个月至2岁1个月期间共间断呕吐发作4次,伴腹泻、嗜睡、昏迷及肝损害,为进食水果后或上呼吸道感染后发作,每次发作时经葡萄糖、碳酸氢钠等静脉输注后病情很快缓解。发作间期无异常。患儿于2岁5个月时进食较多水果30 min后呕吐、昏迷、抽搐,伴发热。当地医院发现低血糖(0.52~2.30 mmol/L),代谢性酸中毒,酮症,肝损害,脑水肿。经治疗后一般情况好转,仍间断抽搐,四肢痉挛型瘫痪。基因分析显示患儿FBP1基因存在c.974T>A和c.755A>T 2个突变,父母各携带1个突变,均为未报道的新突变,证实为果糖-1,6-二磷酸酶缺乏症。
果糖-1,6-二磷酸酶缺乏症为罕见的糖异生障碍性疾病,诊断困难,主要治疗方法为限制果糖、避免长期饥饿、感染等疾病,急性期补充葡萄糖溶液,多数预后良好。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
果糖-1,6-二磷酸酶缺乏症(OMIM 229700)为一种罕见的常染色体隐性遗传代谢病,由FBP1基因突变导致。果糖-1,6-二磷酸酶为糖异生途径的关键酶,催化1,6-二磷酸果糖水解为6-磷酸果糖及磷酸盐[1,2]。FBP1基因定位于染色体9q22.2-q22.3,由7个外显子组成,共含有约31 000个碱基对[3]。患儿在大量摄入果糖、长时间饥饿或急性疾病状态下诱发糖异生障碍,出现急性代谢紊乱,导致酮症性低血糖、高乳酸血症、代谢性酸中毒、肝病、脑病等并发症[4]。果糖-1,6-二磷酸酶缺乏症发病率极低,法国报道低于1/900 000[5],我国罕有文献报道[6]。现就1例因果糖-1,6-二磷酸酶缺乏症导致的严重脑病、肝病患儿的诊疗经过及基因突变进行研究,了解本病的发病过程及其基因突变的特点,并进一步扩展本疾病的基因突变谱。
患儿,男,2岁5个月时因"间断呕吐、腹泻、昏迷及嗜睡1年余"就诊。患儿1岁3个月内发育正常,1岁3个月时进食大量甜瓜,1 d后呕吐、腹泻、腹痛,当地医院考虑肠套叠,经气灌肠治疗后好转。平素厌食水果及甜食,自述吃水果后腹痛,喜欢肉类、蛋、粮食。患儿1岁3个月~2岁1个月间断呕吐发作4次,伴腹泻、嗜睡、昏迷及肝损害,进食水果后或患上呼吸道感染时发作,伴酸中毒,血pH低至6.9,未检测血糖,每次发作时经葡萄糖、碳酸氢钠等静脉输注后病情很快缓解。外院血氨基酸、酯酰肉碱谱及尿有机酸分析结果大致正常。发作间期无异常。患儿于2岁5个月时进食较多水果,30 min后出现呕吐,昏迷,抽搐。当地医院检测发现低血糖(0.52~2.30 mmol/L,正常参考值3.61~6.11 mmol/L),代谢性酸中毒,血乳酸增高(2.2~15.9 mmol/L,正常参考值0.5~2.0 mmol/L)。尿酮体阳性,尿蛋白微量。血清电解质正常,丙氨酸转氨酶(ALT)增高(78~123 IU/L,正常参考值0~40 IU/L),天冬氨酸氨基转移酶(AST)增高(94~154 IU/L,正常参考值0~45 IU/L)。头颅CT、磁共振成像(MRI)显示弥散性脑水肿、尾状核、苍白球及壳核肿胀(图1、图2),可见广泛血管炎性水肿及细胞毒性水肿,符合低血糖脑病。静脉滴注葡萄糖、碳酸氢钠、甘露醇及保肝药物后一般情况好转,但此后睡眠中时有抽搐,为局灶性发作,四肢肌张力增高,伴痉挛型瘫痪。
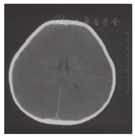
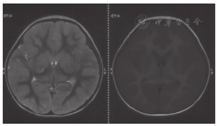
出生史及家族史:患儿系第4胎,第4产,足月顺产出生,出生体质量3 900 g。母乳喂养至1岁,此后混合喂养。2岁5个月前智力运动及体格发育正常,按计划接种疫苗。父母健康,为非近亲婚配。3个同胞姐姐健康,无偏食倾向。
体格检查:神志清楚,精神反应正常,竖头不稳,体质量12.5 kg,身长80.0 cm,头围48.5 cm。肤色正常,呼吸均匀,双肺及心脏查体未见异常,肝脏肋下未触及,四肢肌力Ⅴ级,放松状态下肌张力正常,刺激后肌张力显著增高,躯干呈角弓反张状,双侧膝腱反射亢进,双侧巴氏征(+)。
实验室检查:血常规结果正常。尿蛋白(阴性~微量),尿糖(-),尿酮体(-)。血糖(4.9 mmol/L)、血清电解质、肝功能、肌酸激酶、乳酸、β羟丁酸、血氨正常。血清维生素D降低(12.6 μg/L,正常参考值30~40 μg/L)。尿液乳酸增高,酮体及甘油酸增高,其余有机酸正常。心电图及超声心动图未见异常。腹部超声显示右侧肾盂扩张,肝略大,胆囊壁水肿增厚,双肾、输尿管未见明显异常。视频脑电图显示异常小儿脑电图,背景慢活动。
结合患儿病情经过及饮食特点,临床诊断为低血糖脑病,疑似果糖代谢异常。为明确病因,采用新一代基因测序技术进行基因分析(北京福佑龙惠遗传病专科门诊部协助)。对于患儿父母及正常对照DNA样本,采用一代测序方法进行验证。结果显示:患儿FBP1基因检出c.974T>A(p.V325E)和c.755A>T(p.D252V)2个突变,为复合杂合突变,符合果糖-1,6-二磷酸酶缺乏症(图3)。2个突变均为未报道的新突变,c.974T>A来自父亲,突变导致果糖-1,6-二磷酸酶多肽链第325位氨基酸由缬氨酸改变为谷氨酸。c.755A>T来自母亲,导致该多肽链第252位由天冬氨酸改变为缬氨酸。
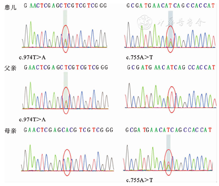
基因突变致病性分析:在正常对照及人类基因突变数据库(HGMD)及千人基因组计划资料库(www.1000genomes.org)中未检出患儿FBP1基因c.974T>A和c.755A>T 2个突变。经Mutation Taster及UCSC网站致病性预测及多物种同源性分析,c.974T>A及c.755A>T突变位点在哺乳动物中高度保守,均提示可能致病。
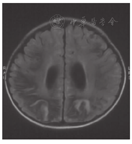
饮食治疗原则为限制果糖摄入,避免食用水果、奶类、南瓜、动物肝脏及胡萝卜,鼓励肉、蛋、豆类、鱼虾、粮食及菜类等食物,避免长时间饥饿。为缓解肌紧张,口服盐酸苯海索片及氯硝西泮。为保证营养,给予左卡尼汀、维生素B12、A、D等支持治疗。治疗后1个月后复查脑MRI显示侧脑室扩张,提示脑萎缩(图4)。随访至3岁,智力逐渐恢复,痉挛型截瘫逐渐好转,治疗后未再出现呕吐、腹痛及抽搐等症状。
果糖-1,6-二磷酸酶缺乏症为罕见的严重糖代谢障碍性疾病,由于1,6-二磷酸果糖水解为6-磷酸果糖及磷酸盐的过程受阻,患儿在进食大量果糖、长时间饥饿或急性感染时糖异生障碍,引起低血糖、酮症及乳酸酸中毒等代谢危象发作,导致脑、肝损害,严重致死[5]。
Baker和Winegrad[7]于1970年首次报道本病,迄今全球已报道100余例[1,2,6,7,8]。患者临床表现缺乏特异性[9],多于长时间饥饿、发热、进食大量果糖或饮酒后出现急性代谢紊乱,亦可进食百里香后起病[10]。患儿多于2岁内起病[5],其中约50%为出生后1~4 d出现症状,因严重乳酸酸中毒及低血糖导致过度通气。最早于出生后数小时发病,表现为新生儿低血糖症,由于糖原贮存不足,预后多数不良。亦可晚至成年期发病[1],并逐渐出现易激惹、嗜睡、昏迷、呼吸困难、心动过速、肌张力低下及肝大等症状。本例患儿的临床表现与国外报道病例类似,在进食大量水果及急性感染后出现酮症性低血糖及乳酸酸中毒,遗留痉挛型截瘫等神经系统后遗症,既往文献中未见有患儿相关影像学报道,本研究报道了果糖-1,6-二磷酸酶缺乏症低血糖脑病后继发性痉挛型瘫痪患儿的头颅影像学变化。
果糖-1,6-二磷酸酶缺乏症患儿典型的实验室改变为酮症性低血糖及乳酸酸中毒,乳酸/丙酮酸比值可增高至30倍以上,一些患者伴肝损害、胰高血糖素抵抗性低血糖或暂时性高三酰甘油血症[11],血清游离脂肪酸及尿酸水平增高。尿有机酸分析可见乳酸、酮体、甘油及3-磷酸甘油等糖异生底物水平增高[9,11,12]。本例患儿多次发病急性期可见酮症性低血糖症及乳酸酸中毒,伴肝损害、高乳酸血症及酮症,符合果糖-1,6-二磷酸酶缺乏症特征。
1995年El-Maghrabi等[3]首次报道FBP1基因的结构及定位,迄今已报道35种突变[4],最常见的突变为c.959dupG(c.960_1insG)[2,5,6,13],可为纯合或杂合突变。c.959dupG在日本人群中发病率最高,Kikawa等[2]曾报道该位点的发生率高达46%。c.841G>A为另一常见突变[4,11],此外,也可见c.685C>T[5]及c.658delT[14]等突变。本例患儿FBP1基因2个新突变c.974T>A和c.755A>T,分别来自父母,在不同物种间存在高度保守性,经过mutation taster及UCSC Genome Browser等软件预测2个突变均极可能致病。
既往曾报道果糖-1,6-二磷酸酶缺乏症患儿中,并未发现FBP1致病突变[2],可能存在FBP1基因启动子或编码控制2,6-二磷酸果糖水平的双功能酶相关基因突变[6,9]。体外培养的外周血淋巴细胞或单核细胞酶活性检测同样可以为该疾病的诊断提供依据[15,16],果糖-1,6-二磷酸酶活性可下降至正常值的30%以下[9],严重者酶活性测定值可低至0。但外周血淋巴细胞酶学活性检测在正常范围内同样不能除外本病[16]。患儿发作间歇期多无明显临床症状,部分患儿可通过果糖、甘油或丙氨酸负荷试验及饥饿试验诱发出急性症状,因为诱发试验存在一定危险性,所以不宜临床推广[9]。
对于疑诊果糖-1,6-二磷酸酶缺乏症患儿,应开始静脉滴注或口服葡萄糖。对于严重低血糖患儿,应静脉推注200 g/L葡萄糖溶液,而后快速持续静脉滴注葡萄糖,并给予碳酸氢钠,以纠正低血糖症及酸中毒。对于本病患儿须严格禁止输注甘油或果糖,对于脑水肿患儿应予以注意[17]。维持期治疗原则为限制果糖、蔗糖及山梨糖摄入,避免长时间饥饿,急性期补充足够葡萄糖,可增加喂养次数,食用生玉米淀粉,必要时应用胃管持续喂养。严重代谢危象时,可适当限制脂肪及蛋白质摄入。在疾病间歇期,不需要额外补充葡萄糖。
果糖-1,6-二磷酸酶缺乏症急性发作期凶险,因低血糖症及顽固性酸中毒可能导致死亡[5],存活患儿可能遗留瘫痪、癫痫、智力运动障碍等后遗症。经正确治疗,可有效避免代谢危象的发生。经饮食及生活干预后,多数患儿转归良好,体格及智力运动发育同健康同龄儿。患儿对饥饿的耐受力随年龄增长而逐渐提高[9],成年后可正常结婚生育[18]。本例患儿确诊后随访7个月,现在3岁,智力逐步恢复到病前水平,痉挛型瘫痪也在逐渐恢复过程中,未再出现呕吐等发作。
果糖-1,6-二磷酸酶缺乏症是遗传代谢病中少数可以治疗的疾病,急性发作期疾病凶险,病死率及致残率很高。建议临床医师对于不明原因的低血糖、代谢性酸中毒患儿,高度重视潜在代谢病的筛查,如果尿有机酸分析、血液氨基酸及酯酰肉碱谱分析正常,需及早进行基因分析,明确病因。

























