
人类芳香化酶缺乏症是一种罕见的先天性雌激素缺乏综合征,是由编码芳香化酶的基因(CYP19A1)失功能突变所致。现对芳香化酶基因的结构和组织表达,芳香化酶在人体雌激素合成中的关键作用及芳香化酶缺乏症男性和女性患者的临床表现、诊断和治疗进行阐述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
芳香化酶缺乏症(aromatase deficiency,AD)是芳香化酶基因(CYP19A1)失功能突变所致的一种罕见的先天性雌激素缺乏综合征。现总结近30年来的相关文献,对芳香化酶基因的结构和组织表达,芳香化酶的重要生理作用及AD的临床表现、诊断和治疗进行阐述,以期提高广大临床医师对AD及相关疾病的诊治水平。
P450芳香化酶(P450 aromatase,CYP19A1;OMIM 107910;GeneID 1588)于1988年从胎盘中分离[1,2],其催化雄烯二酮、睾酮(T)和16 α-羟雄烯二酮分别转化为雌酮(E1)、雌二醇(E2)和雌三醇(E3)[2]。人类芳香化酶基因—CYP19A1(cytochrome P450,family 19,subfamily A,polypeptide1)定位于染色体19q21.1,含有10个外显子:外显子1与基因组织特异性表达有关,但不参与蛋白编码;外显子2~10编码由503个氨基酸组成的芳香化酶蛋白(图1)[3,4,5,6]。特异性启动子决定CYP19A1基因的组织特异性表达(图1)[2,6];由于编码区相同,各种组织合成的芳香化酶只有一种而且功能相同[2]。
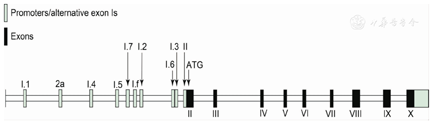
芳香化酶在多种组织的细胞内质网膜上表达,如卵巢的粒层细胞和黄体,睾丸的Leydig细胞和Sertoli细胞,乳房,胎盘的合胞体滋养层,神经元,脑(包括下丘脑),肝脏,前脂肪细胞,成纤维细胞,血管平滑肌细胞,软骨细胞和成骨细胞等[7]。
芳香化酶是人体雌激素合成的关键酶,其将雄激素底物雄烯二酮、T和16α羟雄烯二酮分别转换为E1、E2和E3[7]。怀孕期间,胎儿肾上腺和母体肾上腺产生大量的硫酸脱氢表雄酮(DHEA-S),在胎儿肾上腺和肝脏,DHEA-S被16α羟基化。循环中来自胎儿的16α羟-DHEA-S和来自母体的DHEA-S被转移至胎盘,其硫酸基团被胎盘硫酸酯酶切割,然后,3β羟类固醇脱氢酶(3βHSD)催化这些类固醇转化为雄烯二酮和16α羟雄烯二酮,17βHSD催化这2种激素分别转化为T和16α羟基睾酮,最后再通过胎盘芳香化酶的作用转化为雌激素(主要是E3)[8](图2)。胎盘芳香化酶保护胎儿免受胎儿雄激素的雄性化作用。
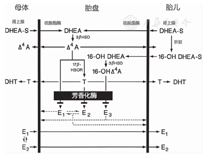
注:DHEA-S:硫酸脱氢表雄酮;3βHSD:3β羟类固醇脱氢酶;DHEA:脱氢表雄酮;Δ4A:雄烯二酮;17βHSOR:17β羟类固醇氧化还原酶;DHT:双氢睾酮;T:睾酮;E1:雌酮;E2:雌二醇;E3:雌三醇 DHEA-S:dehy-droepiandrosterone sulfate;3βHSD:3β hydroxysteroid dehydrogenase;DHEA:dehydroepiandrosterone;Δ4A:Δ4-5-isomerase androstenedione;17βHSOR:17β hydroxysteroid oxidoreductase;DHT:dihydroxytestosterone;T:testoste-rone;E1:estrone;E2:estradiol;E3:estriol
胎盘从由胚泡的滋养外胚层发育而来,在遗传学上属胎儿组织[9]。由于胎儿芳香化酶缺乏,上述雌激素正常合成受阻,使母亲和胎儿暴露于大量的雄激素,导致孕有胎盘AD胎儿的母亲在妊娠期间男性化逐渐加重,女性胎儿出生时外生殖器不同程度地男性化,及其后男性和女性患者在不同发育时期表现出的生殖、骨骼及代谢等系统多种异常临床表现[2,9]。
1991年日本学者首次报道了AD的临床病例[10],次年证实该女性患儿存在芳香化酶基因纯合子突变[11]:5′端的剪接受体序列GT突变为GC,导致位于内含子6下游的隐性剪接受体的启用,结果来自内含子6的87个bp插入到编码区,使成熟的多肽链中多出29个额外氨基酸;其父母为该位点的杂合子突变[12]。
截至2016年,已报道34例AD患者(女23例,男11例)[13]。AD遗传方式为常染色体隐性遗传,发病多见于近亲结婚家庭[14]。发病无种族热点,患者来自日本[10,11,12]、瑞士[2,15,16]、意大利[17,18]、德国[19,20]、阿根廷[21,22]、美国[8,23,24]、澳大利亚[1]、土耳其[25]、印度[26]和中国[13,27]等。至今,已报道的致病突变30多种,包括点突变、缺失、插入和几个剪接位点突变[2,13]。点突变多发生于酶功能重要区域,如外显子5(已报道8种突变[2,16,21,22,24,28],其中1种为复合杂合突变[21])编码酶催化活性所必需的氨基酸;高度保守的外显子9(已报道8种突变[13,15,17,18,21,29,30,31],其中复合杂合突变4种[13,17,18,21])编码底物结合区域,外显子10(已报道4种突变[8,23,26,27],其中1种为复合杂合突变[27])则编码关键的血红素结合区域[7]。目前已报道的芳香化酶外显子3的3种突变均为复合杂合突变[13,15,27],其中2种来自中国[13,27]。对部分突变进行的酶功能学研究显示,有的突变导致芳香化酶活性完全丧失[16],有的则残留不同程度的酶活性(如有报道突变后酶活性只有野生型的0.2%[14,32];有的则为14.3%[27])。
孕有严重芳香化酶缺乏胎儿的母亲在孕期由于体内E1、E2和E3水平显著低于正常水平,同时T、5α-双氢睾酮和雄烯二酮等雄激素水平显著升高[5],导致她们在怀孕3个月后开始出现男性化表现(如痤疮、多毛症、胡须、阴蒂肥大、声音低沉等),并逐渐加重,在分娩时达高峰[13,26]。母亲男性化症状通常在分娩后5~6个月消退,也有迟至22个月后才消退,但仍有声音低沉[2,15,16]。有些母亲孕期不出现男性化[2,13,23,27],可能是由于胎儿肾上腺分泌的雄激素低于正常和/或由于残留的胎盘芳香化酶活性使胎盘产生足够雌激素以拮抗增多的雄激素作用[27]。目前有学者认为,低至1%的芳香化酶活性即可保护母亲不发生严重的男性化[33],如芳香化酶纯合子突变(R192H)的兄妹俩,功能研究显示突变后该酶的催化活性为野生型的19%(﹥1%),其母孕期无男性化表现[2]。
女性患儿(染色体检查为46,XX)出生时女性假两性畸形、外生殖器男性化;儿童时期肥胖、高身材;青春期发育落后,高促性腺激素性性功能减退、多囊卵巢、骨龄成熟落后及高胰岛素血症等[13,26]。影响这些临床表现的因素有年龄、性别和酶活性水平[13]。
女性患儿第一个突出临床表现是出生时外生殖器不同程度的男性化[28],70%的女性患儿表现为严重男性化(Prader分期Ⅳ或Ⅴ期)[2],这提示患儿在宫内暴露于高雄激素环境,后者也影响卵巢功能,未来行为的大脑程序化,成年期脂肪和糖代谢及心血管系统代谢的程序化[2]。严重的生殖器模糊则提示女性胎儿外生殖器在孕第12周前接触大量T和双氢睾酮[9]。由于卵巢芳香化酶缺乏,在青春期女性患儿男性化继续发展,缺乏女性第二性征的发育和青春期生长加速,促性腺激素升高,雄激素及其前体在体内积聚,导致男性化进展、卵巢过度刺激及多囊卵巢[2,14]。由于中枢促性腺激素反馈的再设定,导致血卵泡刺激素(FSH)中度甚至显著升高,促黄体生成素(LH)轻度升高[28],而FSH和LH水平的异常早在胎儿期及新生儿期就已存在[13]。长期促性腺激素分泌过多导致卵巢囊肿[2,31],有报道1例女性患儿出生5个月时即发现存在双侧卵巢囊肿(4 cm×2 cm)[21]。1例女性患者18岁才就诊,给临床提供了AD的自然病史:14岁时无乳房发育,原发性闭经,阴蒂进行性增大,阴毛Tanner 3期,雄激素增高,雌激素检测不到,超声检查显示双侧卵巢有多个4~6 cm大小的囊肿[23]。雌激素治疗使促性腺激素降低,乳房发育,青春期生长加速,月经来潮和卵巢囊肿回缩[23]。芳香化酶活性部分残留足以不断合成雌激素刺激乳房和子宫发育[2],临床上有些女性患者可有自发乳房发育[28,34,35],自发初潮并随后有规则月经周期[28]。有的女性患儿血促性腺激素水平在青春期正常范围,卵巢大小正常[31]。
与女性患儿不同,男性患儿出生时外生殖器正常,多数在婴儿及儿童期无症状[2]。通常在成年早期(24~31岁)因高身材和代谢异常才被诊断[2]。只有1例因母亲孕期男性化而于出生后不久被诊断[16]。多数男性患者T正常偏高或明显升高伴FSH升高。雄激素升高的原因是芳香化酶缺乏导致雄激素转化为雌激素减少,升高的LH刺激Leydig细胞合成雄激素过多[32]。少数男性患者可有生殖器畸形(如尿道下裂[2],双侧或单侧隐睾[2,18,22]),其与AD的关系尚有待阐明[2]。也有巨大睾丸[32](macroorchidism,体积为34 mL)的病例报道,可能是异常升高的FSH作用于正常的睾丸所致[14],雌激素治疗后睾丸体积降至28 mL[32]。此外,少数患者的精液分析或睾丸活检显示精子发生障碍,少精子症、弱精子症,严重者可导致女方不孕[17,19,29]。
多数男性患者就诊延迟,就诊时平均年龄为25.8岁(22~31岁),骨龄为15.1岁(14.0~16.5岁),骨龄落后10.8岁(7.0~16.5岁)[19]。由于骨龄闭合延迟和生长期延长,多数男性患者成年期身材高大,身高均数为191.8 cm(182.5~204.0 cm)[14,16,18,19,22,27,29]和类阉者体型。
多数AD患者有广泛骨疼痛,在成年期仍有持续和渐进的线性生长,骨成熟延迟,骨骺未闭合,类阉者体型,渐进膝外翻,骨量减少、骨质疏松症和骨密度(BMD)降低等[36]表现。这主要与雌激素缺乏相关,研究表明芳香化酶在骨骼(主要是成骨细胞和软骨细胞)中广泛表达,雌激素直接参与骨成熟、BMD的获得和维持等[37]。先天性雌激素缺乏通过延迟骨峰值发生而不是通过骨质吸收机制导致骨质减少和骨质疏松等[37]。骨量峰值达到后,还需要雌激素来维持骨量[38]。因此,临床上几乎所有AD男性患者表现出典型的特征:骨骺未闭合、骨质减少、骨质疏松和类阉者体型[39]。
膝外翻的病因尚不清楚,可能由于下肢骨骺未闭合、骨密度下降和膝关节退行性变化等导致力学的改变[18,19,27,29];腹型肥胖也可增加患者膝关节的负重,引发骨骼畸形[27]。
有身高数据的8例女性患者中,6例身高高于其所在人群平均身高(身高SDS:+1.44~+3.50)[1,13,14,21,26,40],但有2例身高低于人群平均身高(身高SDS分别为-0.91、-1.53)[8,25],骨龄落后平均为-2.3岁(-0.7~-5.0岁)[1,8,13,14,21,25,26,40]。而BMD从降低[14,26]至处于正常范围[21,40]均有报道。2014年报道了直到成年期(25岁)因骨折就诊才被诊断的1例AD女性患者[1],给临床提供了AD对女性患者骨骼系统影响的自然病史,其临床表现为骨质疏松、身材高大(180 cm,靶身高161 cm)等[1]。
雄激素过高和雌激素过少对代谢影响是AD的重要特征[18],其表现与代谢综合征症状相似,男性患者尤其明显,表现为腹型肥胖,多数男性患者体质量指数(BMI)>25 kg/m2,高胰岛素血症、胰岛素抵抗、黑棘皮病,非酒精性脂肪性肝炎,2型糖尿病与早期颈动脉脉粥样硬化,并通常伴随着三酰甘油(TG)和低密度脂蛋白(LDL)升高,高密度脂蛋白(HDL)降低,曲线下胰岛素面积与C肽升高及高血糖[14,18,19,22,24,27,41]。1例成年男性AD患者在诊断前接受高剂量T治疗引起雌激素和雄激素的比例严重失衡而导致胰岛素抵抗和2型糖尿病[22]。男性患者雌激素治疗后,这些临床表现和实验室代谢异常得到改善,但体质量超重或肥胖不能得到好转[18]。
目前报道的有代谢紊乱表现的AD女性患者较少:1例女性患儿14岁时被检出有轻度血脂异常[42];另1例9岁时开始出现胰岛素抵抗和糖耐量异常,同时伴有血T、雄烯二酮和FSH升高[34]。雌激素治疗不能抑制促性腺激素、T和改善胰岛素抵抗;在加用二甲双胍治疗期间患儿发生2型糖尿病、黑棘皮症及轻度高TG血症;促性腺激素释放激素类似物(GnRHa)治疗1年后血促性腺激素和T水平明显降低,但胰岛素抵抗仍持续存在[34]。提示女孩胎儿期雄激素升高和/或雌激素缺乏可导致胎儿性腺轴及胰岛素敏感性的程序化机制的持久改变[34]。
由此可见,男性和女性AD患者在临床上都有发生代谢综合征的风险,但发生代谢综合征的男性AD患者报道多于女性,可能是由于女性患者更易被早期发现并在青春期前开始雌激素治疗,因而由雌激素缺乏引起的严重、慢性代谢异常很可能未能表现出来。动物研究也发现,芳香化酶基因敲除(ArKO)小鼠随年龄的增大逐渐出现糖耐量异常和胰岛素抵抗,雌激素治疗可改善这种糖代谢异常[43]。上述临床及动物实验研究显示雌激素在维持糖代谢稳定方面起重要作用。
首先了解患者的相关病史:母亲孕期有无男性化,父母是否近亲婚配;女性患者或男性患者的姐妹在出生时是否外生殖器性别模糊或男性化,是否有青春期发育落后;患者父母及患者兄弟姐妹的身高以判断患者身高是否异常增高;患者从出生到成年期的生长发育情况,有无隐睾病史等[36]。
对母亲孕期有男性化,女性患儿出生时有外生殖器性别模糊或男性化;婴儿期或青春期前男童其家庭成员中有AD病史的,可直接进行遗传分析;对其他患者需根据病史和相关临床特征,结合性激素和骨龄等检测结果,对临床拟诊的,如E2明显降低或检测不到及骨骺未闭合,T降低或正常,LH正常或升高,FSH明显升高则强烈怀疑芳香化酶缺乏[36],需再进行遗传分析以明确诊断。
遗传分析:由于根据AD的症状、体征、激素和骨龄检查等做出的临床诊断可靠性差,对临床拟诊患者需进行CYP19A1基因分析[36]以明确诊断:对CYP19A1(外显子2~10)DNA测序;在转染细胞中进行mRNA表达分析和芳香化酶活性分析;标准核型分析及对患者家族成员的遗传分析[36]。
对明确诊断的患者还需要做代谢、骨和生殖系统等方面的相关检查:(1)影像学检查,包括双能X线吸收测量法(DEXA)测量骨密度(L2~L4和股骨颈)和肝脏超声检查;(2)血生化检查,空腹血糖及胰岛素水平、血脂、肝功能、骨转换指标、胰岛素样生长因子-1(IGF-1)、性激素结合球蛋白(SHBG)和DHEA-S水平及男性患者的精子分析等[36]。
(1)母亲孕期男性化:由于怀有芳香化酶缺乏胎儿的孕母血E2、E3水平极低,可与母体卵巢肿瘤或母体摄入雄激素等导致孕母男性化的疾病相鉴别[5]。(2)出生时女婴外生殖器性别模糊或男性化:出生时女婴外生殖器男性化的原因有21-羟化酶缺乏,其他类型的先天性肾上腺皮质增生症(CAH),母亲摄入雄激素类物质,母亲患有合成雄激素的肿瘤和AD[25]。AD虽然是女性胎儿男性化的少见病因,在XX性发育异常的鉴别诊断中应包括AD[20],新近有AD女性患儿被误诊为CAH的报道[25]。女性外生殖器性别模糊需要鉴别诊断的疾病:导致男性化的CAH(如21-羟化酶缺乏、11β羟化酶缺乏、3β HSD缺乏),真两性畸形,经胎盘的致男性化的药物(雄激素类和孕酮)等[44]。(3)其他需鉴别的疾病:血清性激素和促性腺激素水平的测定有助于AD与其他疾病鉴别(表1)。雌激素抵抗与AD具有相同的临床特点,但由于其血清E2水平明显高于正常,易于诊断(表1)。
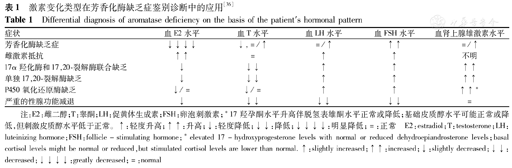
激素变化类型在芳香化酶缺乏症鉴别诊断中的应用[36]
Differential diagnosis of aromatase deficiency on the basis of the patient′s hormonal pattern
激素变化类型在芳香化酶缺乏症鉴别诊断中的应用[36]
Differential diagnosis of aromatase deficiency on the basis of the patient′s hormonal pattern
| 症状 | 血E2水平 | 血T水平 | 血LH水平 | 血FSH水平 | 血肾上腺雄激素水平 |
|---|---|---|---|---|---|
| 芳香化酶缺乏症 | ↓↓↓↓ | ↓,=/↑ | =/↑ | ↑↑ | =/↑ |
| 雌激素抵抗 | ↑↑ | = | ↑ | ↑ | 不明 |
| 17α羟化酶和17,20-裂解酶联合缺乏 | ↓ | ↓↓ | ↑ | ↑ | ↑↑ |
| 单独17,20-裂解酶缺乏 | ↓ | ↓↓ | ↑ | ↑ | ↑↑ |
| P450氧化还原酶缺乏 | ↓/= | ↓/= | ↑ | ↑ | ↑↑* |
| 严重的性腺功能减退 | ↓ | ↓↓ | ↓↓ | ↓↓ | = |
注:E2:雌二醇;T:睾酮;LH:促黄体生成素;FSH:卵泡刺激素;*17羟孕酮水平升高伴脱氢表雄酮水平正常或降低;基础皮质醇水平可能正常或降低,但刺激皮质醇水平低于正常。↑:轻度升高;↑↑:升高;↓:轻度降低;↓↓:降低;↓↓↓↓:明显降低;=:正常 E2:estradiol;T:testosterone;LH:luteinizing hormone;FSH:follicle-stimulating hormone;*elevated 17-hydroxyprogesterone levels with normal or reduced dehydroepiandrosterone levels;basal cortisol levels might be normal or reduced,but stimulated cortisol levels are lower than normal.↑:slightly increased;↑↑:increased;↓:slightly decreased;↓↓:decreased;↓↓↓↓:greatly decreased;=:normal
临床表现(身材高大、骨龄延迟、骨质疏松和类阉人样骨骼)与AD相似的其他疾病[36]:17α羟化酶和17,20-裂解酶联合缺乏(combined 17α-hydroxylase-17 and 20-lyase deficiency),单独17,20-裂解酶缺乏(isolated 17,20-lyase deficiency)、P450氧化还原酶缺乏(P450 oxidoreductase deficiency,PORD)和青春期前严重的性腺功能减退。这些疾病都表现为雌激素相对缺乏及更多其他临床特征,如生殖器低雄激素化,而AD缺乏这些特征。17α羟化酶和17,20-裂解酶联合缺乏、单独17,20-裂解酶缺陷均有血雄激素明显降低,雌激素降低[36,45];17α羟化酶和17,20-裂解酶联合缺乏由于去氧皮质酮(DOC)水平异常升高可导致高血压和低血钾等[45],结合其他临床特征可与AD鉴别。根据缺乏肾上腺功能不全、明显骨骼发育异常和性发育异常等临床特征,可除外PORD[46]。如患儿青春期发育正常及血性水平正常可排除性腺功能减退。
由于Klinefelter综合征的一些临床特征(如高身材、类阉者体型)与AD相似,需进行核型分析以除外其诊断及其他染色体异常[36]。
雌激素治疗目的:在儿童早期维持BMD和预防发生卵巢囊肿;其后促进乳房发育,诱导生长加速和骨骺闭合[15]以及在青春期相应的时间开始并维持月经周期[47]。目前,女性AD患者应用雌激素预防治疗雌激素缺乏的经验有限,关于雌激素开始治疗的年龄和起始剂量也无一致的观点。开始连续口服低剂量结合雌激素,然后根据临床对治疗的反应、患者的年龄和发育阶段等适当调整。低剂量雌激素(50~100 μg/d)为儿童早期的正常线性生长和骨龄成熟所必需;而青春期前及青春期儿童垂体-卵巢轴负反馈及乳房发育等则需要较高剂量的雌激素(1.4~2.0 mg/d)[40]。其后加用孕激素诱导月经初潮。
目前的治疗经验主要来源于9.6~14.0岁的不能启动青春期发育的女性患儿[13]。在雌激素治疗的8例患儿中,有出现正常的青春期生长加速(6例)、乳房发育(7例)和月经初潮(3例)[13]。多数患儿治疗后出现青春期生长加速、乳房发育[13]。但只有2例患儿男性化体征消失,3例骨龄落后改善。雌激素治疗还可导致血促性腺激素水平和雄激素水平降低,卵巢囊肿消退。只有1例患儿于儿童早期(3.5岁)开始低剂量雌激素治疗直至青春期(15岁),长期随访发现该患儿达到接近成年身高,青春期发育和骨骼成熟正常[40]。对该患儿进行长期雌激素治疗发现,低剂量雌激素(0.05~0.10 mg/d)为儿童早期正常生长和骨骼成熟所必需。停止治疗则导致骨骼成熟停滞,生长速度降低[40];治疗使其达到正常成年身高[174.5 cm;+2.4 SDS,在其靶身高上限(169.6±8.5) cm]、正常青春期发育和BMD,但患儿仍有多囊卵巢,尤其在青春期,提示这种低雌激素剂量不足以抑制促性腺激素的分泌,有效抑制垂体-卵巢轴则需要更大剂量的雌激素[40]。
治疗期间应监测血促性腺激素水平和卵巢大小[13]。在整个儿童期和青春期需调整雌激素的治疗剂量,以保证骨骼正常发育,青春期生长加速,正常BMD的获得以及在适当年龄开始女性第二性征的发育[40]。但有的患儿促性腺激素水平,特别是FSH不能被适当抑制[13]。
在成年后才开始治疗的患者中代谢综合征的表现比较突出,雌激素治疗后可使轻度血脂异常得到纠正,但高胰岛素血症仍存在。雌激素替代治疗后,有些患者雌激素缺乏症状,如FSH升高、多囊卵巢影像学变化未能改善[28],这可能与外周组织,如下丘脑、卵巢局部从雄激素前体合成雌激素缺乏有关[28]。
目前已报道的AD男性患者多数在成年期才被诊断,对于这些成年患者,雌激素治疗目的是促进骨骼成熟、骨骺闭合,使BMD恢复正常及纠正雌激素缺乏引起的代谢异常[13,36]。高剂量雌激素可使AD成年男性的骨骼成熟在6~9个月完成[19,29],并可防止患者身高进一步增高和类阉者体型的加重[36]。雌激素治疗AD患者可增加其BMD,使其在6~9个月恢复正常[24,36],并可使促性腺激素分泌正常化,血脂水平降低,改善糖代谢、胰岛素敏感性和肝功能,进而改善患者的代谢异常及降低心血管风险[19,36,48,49]。但成年期开始治疗不能改善患者的生精受损、脂肪分布异常、肥胖和骨骼畸形等。在实现骨成熟、骨骺闭合和骨矿化正常化等目标后,作为一种终身替代疗法,应继续雌激素治疗,其主要目的是防止骨质流失,减少心血管疾病的风险[36]。对于在出生时或婴儿期已明确诊断的男性患儿,应在青春期开始时给予低剂量E2[0.80~0.12 μg/(kg·d)]治疗,从青春期中期(16~17岁)逐渐增加剂量,以模仿正常男孩青春期发育过程中血清E2水平的持续、逐渐升高[36]。青春期前应避免使用高剂量E2治疗,因为其能抑制青春期生长突增[36]。有学者提出使雌激素水平正常并维持正常骨量的经皮雌激素的适合剂量为每周50~75 μg,分2次使用[48]。较大剂量雌激素治疗可使患者雄激素水平低于正常,因此有报道在治疗6个月后将雌激素剂量减少到12.5 μg,每周2次,可使雄激素水平正常,并可维持正常的FSH、LH和雌激素水平[19]。
共轭雌激素、戊酸雌二醇、雌二醇贴片和雌二醇凝胶等均可有效治疗男性患者[36]。剂量取决于剂型和给药途,优选含有E2的雌激素制剂,因为这种激素成分易于临床血清标本的检测[36]。
在治疗期间应监测雌激素治疗的安全性,避免长期应用超生理剂量雌激素。有效的临床监测指标:BMD、血清E2、促性腺激素(尤其是LH)和T[36]。当这些指标维持在正常范围时提示雌激素剂量适当[50]。骨龄可用于短期和中期雌激素治疗的监测。目前报道的病例中多数开始治疗时患者的骨龄为14岁5个月~16岁5个月,如果在治疗开始后6~9个月骨骺闭合(骨成熟完成)提示雌激素治疗剂量适当[36]。有1例患者开始治疗时骨龄较小(12岁),雌激素治疗41个月后才骨骺闭合[16]。
芳香化酶基因失功能突变所致的AD可导致孕有严重芳香化酶缺乏胎儿的母亲孕期男性化,女性患儿出生时外生殖器假两性畸形和男性化;青春期发育落后、无第二性征发育,原发性闭经、卵巢囊肿增大和男性化进一步加重。男/女患者骨骼成熟延迟、骨龄落后、骨质疏松,缺乏青春期生长加速,成年期继续线性骨生长,身材高及腹型肥胖、高胰岛素血症、血脂异常等代谢紊乱表现。临床医师应提高对该病的认识,对孕期出现男性化母亲所生的所有儿童进行AD筛查[36],有利于AD的早期诊断和及时治疗,从而减轻或改善本病的相关临床表现。从近30年报道的AD患者的各种临床表现中,人们认识到正常雌激素水平在预防胰岛素抵抗和代谢综合征,调节促性腺激素的分泌,防止女性多囊性卵巢相关异常等方面起着重要作用,E2(而非T)不仅为女性,同样为男性青春期生长加速、骨骺闭合及BMD维持正常所必需[51]。

























