
囊性纤维化(CF)是一种危害儿童健康的常染色体隐性遗传病,可累及呼吸、消化、生殖等多系统,预后极差。CF的发生存在种族特异性,欧美人群多见,亚洲人群罕见。近年随着医学技术的进步,越来越多的中国儿童CF患者被发现,可能由于对CF的认识不足,中国CF的发生率被低估。现从发病机制、临床特点、诊断、治疗等多个方面对CF进行介绍,以提高儿科临床医师对本病的关注及认识,做到早期识别与干预,最终改善CF患儿的预后,延长其生存周期。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
囊性纤维化(cystic fibrosis,CF)于1949年被首次报道,是一种累及多系统的常染色体隐性遗传单基因病,可表现为慢性肺疾病、生长发育迟滞、营养不良、脂肪泻、肝脏疾病等,危害儿童健康,影响患儿寿命[1]。CF患儿预后极差,如不治疗,预期寿命不超过10岁。CF的新生儿发病率因国家、种族而异,高加索人发病率最高,为1/25 000~1/1 800,美国白种人为1/3 000,西班牙裔美国人为1/10 000~1/4 000,非洲裔美国人为1/20 000~1/15 000,亚洲人的发病率很低[2,3,4],日本CF发病率低至1/350 000[5]。而我国由于病例数甚少,尚无CF发病率的相关数据。国内学者回顾分析1974年1月至2016年12月的文献,仅收集49例确诊为CF的中国患者[6,7]。但近年来随着对该病认识的提高及基因检测技术的发展,我国对CF的诊断也有所增加。
CF由囊性纤维化跨膜传导调节因子(cystic fibrosis transmembrane conductance regulator,CFTR)基因突变所致。CFTR基因最早于1989年由Riordan等发现,该基因位于7号染色体长臂,全长约250 kb,其产物为CFTR,是一种环磷酸腺苷依赖性Cl-通道蛋白,位于细胞膜顶端,通过吸收Na+,分泌Cl-的方式调节离子转运[8]。目前已报道2 000多个CFTR基因突变位点(http://www.genet.sickkids.on.ca/cftr/app),欧美人群中70%以上存在△F508位点突变。但中国的突变基因与欧美人群差异极大。国内学者报道的49例中国CF患者中,32例存在CFTR突变,共33种突变类型,均为少见突变,其中最常见的突变为c.1766+5G>T,占24.138%,其次为c.2909G>A、c.2684G>A、c.2083dupG、c.595C>T,其余具有异型性[6,7],在高加索人群中不常见。依据CFTR合成、结构、功能的异常可将CFTR基因突变分为6类(表1)[8]。在CF患者中,多数存在 Ⅱ 型突变。不同CFTR突变类型可引起不同的临床表型,Ⅰ、Ⅱ、Ⅲ 型突变易引起胰腺功能不全,临床表现较重;Ⅴ、Ⅵ 型突变因部分Cl-通道存在正常功能,临床症状较轻,且胰腺功能大多正常。一些不伴典型CF表现或仅有轻微病变或单系统病变的患者,称为CFTR相关疾病或CFTR相关代谢综合征。
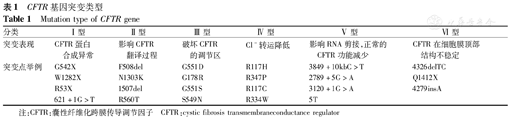
CFTR基因突变类型
Mutation type of CFTR gene
CFTR基因突变类型
Mutation type of CFTR gene
| 分类 | Ⅰ 型 | Ⅱ 型 | Ⅲ 型 | Ⅳ 型 | Ⅴ 型 | Ⅵ 型 |
|---|---|---|---|---|---|---|
| 突变表现 | CFTR蛋白合成异常 | 影响CFTR翻译过程 | 破坏CFTR的调节区 | Cl-转运降低 | 影响RNA剪接,正常的 CFTR功能减少 | CFTR在细胞膜顶部 结构不稳定 |
| 突变点举例 | G542X | F508del | G551D | R117H | 3849+10kbC>T | 4326delTC |
| W1282X | N1303K | G178R | R347P | 2789+5G>A | Q1412X | |
| R53X | 1507del | G551S | R117C | 3120+1G>A | 4279insA | |
| 621+1G>T | R560T | S549N | R334W | 5T |
注:CFTR:囊性纤维化跨膜传导调节因子CFTR:cystic fibrosis transmembraneconductance regulator
CFTR基因合成、结构及功能异常可导致上皮细胞Cl-和水分泌减少,造成黏液堆积,阻塞一些器官的管腔(如呼吸道、胰腺、汗腺等),出现相应的临床症状[9]。呼吸系统常为首要累及的器官,也是最主要的死因(85%)[10]。黏稠的分泌物阻塞支气管,纤毛活动受抑制,而堆积的黏液又可使细菌寄生,持续的炎性反应也会加重肺功能损害,可表现为反复、持续肺部感染、肺不张、气胸、支气管扩张、咯血及呼吸衰竭等[11]。CF患者易并变应性支气管肺曲霉病[12],临床中需要注意。在胰腺,分泌物阻塞腺管,胰酶分泌不足,出现消化不良、营养不良、脂溶性维生素缺乏、生长发育迟滞、脂肪泻、体质量不增、慢性胰腺炎及CF相关的糖尿病等。≥10岁的CF患者均应每年进行空腹血糖及口服葡萄糖耐量试验筛查[13]。肺功能降低和胰腺功能不全所致低体质量与生存率呈负相关,是预后不良的重要因素[14]。在年长儿,小肠分泌功能异常、吸收障碍和胃肠道蠕动减弱可导致低位肠梗阻综合征及慢性便秘。汗氯增高是CF的典型表现,占90%以上[14]。部分病例在新生儿期即可出现症状,如出生后出现胎粪排出延迟、胎粪性腹膜炎、新生儿胆汁淤积、直肠脱垂等,需高度警惕CF的可能。在婴儿期,如出现反复肺炎、吸收不良综合征、局灶性胆汁性肝硬化、低蛋白血症、维生素E缺乏、锌缺乏等,也需警惕CF。此外,在成人中发现,99%的男性CF患者伴先天性双侧输精管缺失导致不育,而女性患者由于宫颈分泌物黏稠导致生育能力下降[14]。
中国49例CF患者临床特点分析结果显示,肺是最常见的受累器官,98%的CF患者有反复呼吸道感染;50%的患者有消化系统症状,依次为胰腺功能不全、肝大、腹泻、黄疸、腹膜炎、肠梗阻等[6,7]。此研究还发现,营养不良、胰腺功能不全在中国CF患者中并不少见,但临床中对此认识不足,因此,对确诊的CF患者应常规进行胰腺功能检查。临床中患儿同时表现呼吸系统和消化系统症状时,特别是出现反复呼吸道感染、支气管扩张及胰腺功能不全时更应警惕CF。
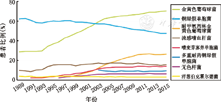
CF患者具有典型的病原学特征,且随年龄变化,病原谱也不同[15]。多数婴儿初始为流感嗜血杆菌和/或金黄色葡萄球菌感染,若无特殊的干预措施,铜绿假单胞菌可最终成为呼吸道感染的主要病原。不同年龄阶段的呼吸道病原分布不同(图1)[16]。2016年美国CF基金会也对不同年份CF的病原变化趋势进行了分析,发现近10年来,金黄色葡萄球菌的感染率明显高于铜绿假单胞菌(图2)[16]。在中国CF患者中,最常见的病原为铜绿假单胞菌,其次为金黄色葡萄球菌、肺炎克雷伯菌等[6]。

典型CF病例可表现为慢性肺部疾病、胰腺功能不全及汗液Cl-升高三联征。CF确诊需结合特有的临床特点、汗液Cl-检测和/或CFTR功能障碍生物化学或遗传标志物检测。2008年,CF基因会确定的CF诊断标准[17]:对有1个或多个临床特征性表现,如慢性、反复性鼻窦或肺部疾病、营养不良和消化道疾病,男性泌尿生殖系统畸形(如输精管缺如)或有CF家族史的患者,首先进行汗液Cl-检测,若2次汗液Cl->60 mmol/L或1次汗液Cl->40 mmol/L及2处CFTR致病突变,则可诊断CF。但2017年,CF基金会发布了CF的最新诊断指南(图3)[18],该指南降低了用于指示疑似诊断CF的汗液Cl-阈值。在所有年龄中,阈值从既往推荐的40 mmol/L降至30 mmol/L。既往研究认为,汗液Cl-在30~39 mmol/L的患者也确诊为CF,而且汗液Cl-正常的患者也可以诊断CF[19]。因此,汗液Cl->30 mmol/L应进一步评估,包括扩展CFTR基因分析和/或CFTR功能分析,以确诊或排除CF。
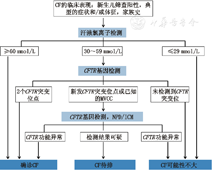
注:CF:囊性纤维化;CFTR:囊性纤维化跨膜传导调节因子;MVCC:临床症状相关的突变;NPD:鼻电位差;ICM:肠电流测量 CF:cystic fibrosis;CFTR:cystic fibrosis transmembrane conductance regulator;MVCC:mutation of varying clinical consequence;NPD:nasal potential difference;ICM:intestinal current measurement
汗液Cl-检测设备价格昂贵,多数地区尚不能进行及时检测,而基因学检测也花费较大,亦不能作为筛查手段,因此,对CF患者的诊断仍然是很大的难题。但近年,研究学者提出了一种新的可用于筛查及诊断CF的方法,即水源起皱试验[20],室温下,将儿童的手浸泡于水中,计时起皱时间,截断值3 min的诊断敏感性为81%,特异性为58%,这种方法操作简单,大大降低了CF患者的筛查及诊断成本。意大利学者对58例CF患者进行研究,结果显示53.4%的CF患者水源起皱试验阳性,提出水源起皱试验是一种进行初筛及提示进行汗液Cl-及基因检测的早期有效方法[21]。对于中国CF患者,由于确诊病例少,且常为罕见突变位点,因此在诊断过程中面临更大的难题,但随着对该病的认识及医学诊断技术的进步,希望能制定适合中国人群的CF诊断流程,从而使更多的中国CF患者得以明确诊断。
目前美国CF中心CF平均确诊年龄为6个月,但也有一些患者在儿童期甚至成年后才确诊,因此整体平均确诊年龄为3岁[2]。而我国由于对CF认识不足,中位确诊年龄延后,在10岁左右[6]。
新生儿筛查对早期诊断CF至关重要,有助于改善CF患儿的营养状况及肺功能。随着新生儿筛查的广泛应用,无症状患儿出生第1个月的诊断率显著提高。在2008年新确诊病例中,43%是通过新生儿筛查而明确诊断的[2]。血片法测定免疫活性的胰蛋白酶原(immunoreactive trypsinogen,IRT)是最早用于新生儿筛查的方法。为避免IRT检测的假阴性,之后研发了IRT/DNA-F508del法,结合CFTR基因主要致病突变位点F508del的检测;随后又研发了IRT/DNA-CFTR多突变检测,包含23~40个CFTR突变的DNA分析,敏感性更高[22]。但由于中国的CFTR突变位点与西方人群差异极大,因此基于欧美CFTR突变谱建立的CF筛查平台对中国患者可能并不适用,急需建立适合中国人群的CF筛查方法。此外,临床医师也应该关注那些临床表现并不典型的中国CF患者。
对CF患者的治疗,需多学科合作、共同干预与指导,并需要规范化的长期管理与随访。这种CF的多学科综合治疗团队模式目前在国外已广泛开展,显著提高了CF患者的生存率,美国CF基金会数据表明,CF患者的预期寿命已从10年前的31岁提高到目前的41岁;英国的一项研究模型预测的CF寿命将达50岁[14]。这种综合的治疗模式值得我国临床医师借鉴与学习。
研究表明,CF患者良好的营养状况与其远期生存及肺功能密切相关[23]。营养支持治疗包括胰酶替代治疗、高热量饮食、脂溶性维生素(维生素A、维生素D、维生素E、维生素K)及矿物质(钠、钙等)补充等。如果患者出现脂肪泻、生长发育落后等胰腺功能不全的表现,应及时补充胰酶,并根据症状调整剂量[24]。但当剂量超过6 000 U/kg时,可导致纤维化结肠病,临床中需注意。如果患者出现营养不良,应积极加强营养,以保证患者肺功能正常。
呼吸道清理有助于CF患者呼吸道黏稠分泌物排出,方法包括体位引流、正压呼气、高频胸壁震荡等[25]。美国CF基金会指南提出,CF患者应自婴儿期起每日进行呼吸道清理[16],但目前尚无研究证实哪种呼吸道清理方法更适合。如果支气管舒张剂可以改善CF患者的肺功能,可以间歇应用。重组人DNA酶可降低呼吸道分泌物的黏稠度,有利于黏液清除,但指南建议用于5岁以上的CF患者。高渗盐水雾化治疗可使纤毛周围层重新水化,改善纤毛-黏液系统的清除功能[26],但在婴儿中尚无研究数据支持,应慎用。
CF患者由于黏液堆积,可使细菌寄生,引起慢性持续感染,尤其是铜绿假单胞菌感染。对初次感染铜绿假单胞菌的患者,一旦发现,即使无临床症状,均应予吸入、口服或静脉抗生素,以杀灭病菌[27]。对慢性铜绿假单胞菌感染患者,吸入抗生素治疗有效,可选择妥布霉素或氨曲南,但当肺部病变严重时,吸入性抗生素不能穿透阻塞的呼吸道,到达大部分受累肺组织,而影响疗效,需要改变给药途径。此外,研究显示,长期使用小剂量大环内酯类抗生素具有抗感染和减少铜绿假单胞菌生物被膜形成的作用,且能够改善患者肺功能,尤其适用于有假单胞菌属定植的患者[28]。但对于无铜绿假单胞菌感染的CF患儿,长期预防性使用抗生素(尤其是全身给药)的疗效尚不明确。
长期感染和炎性反应可引起不可逆的支气管扩张,最终导致呼吸衰竭。终末期治疗仅可选择双肺移植,双肺移植是高风险手术,如何确定最佳手术对象,特别是儿童,目前争议颇大。肺移植指证须符合预测模型要求,即根据第1秒用力呼气容积(FEV1)、年龄、性别、肺部微生物学特征和FEV1下降速率来提示患者是否将面临死亡(如2年之内)。CF患者肺移植指征见表2[29]。肺移植术后5年生存率约为50%,慢性移植排斥反应所致闭塞性细支气管炎是亟待解决的主要问题[30]。

囊性纤维化患者肺移植指征
Index of lung transplantation for cystic fibrosis patients
囊性纤维化患者肺移植指征
Index of lung transplantation for cystic fibrosis patients
| 何时就诊 | ||
|---|---|---|
| FEV1 | 即使给予最佳治疗,FEV1仍快速降低至少30% | |
| 6 min步行路程 | <400 m | |
| 肺动脉高压 | 在无缺氧的条件下:超声示PASP>35 mmHg或右心导管示mPAP>25 mmHg | |
| 随病情加重次数增 | ||
| 多,临床逐渐恶化 | 具备任何以下条件:需要无创通气呼吸支持的急性呼吸衰竭;抗生素耐药率升高且恢复差;尽管营养支持患者营养状况仍恶化;气胸;尽管行支气管动脉栓堵,仍存在威胁生命的咯血症状 | |
| 肺移植时机选择 | ||
| 慢性呼吸衰竭 | 伴低氧血症(<60 mmHg),伴高碳酸血症(>50 mmHg) | |
| 长期无创机械通气 | ||
| 肺动脉高压 | ||
| 经常住院治疗 | ||
| 肺功能急速下降 | ||
| WHO功能分级 Ⅳ 级 | ||
注:FEV1:第1秒用力呼气容积;PASP:肺动脉收缩压;mPAP:平均肺动脉压;WHO:世界卫生组织;1 mmHg=0.133 kPa
FEV1:forced expiratory volume in one second; PASP:pulmonary artery systolic pressure;mPAP:mean pulmonary artery pressure;WHO:World Health Organization;1 mmHg=0.133 kPa
近年来,由于分子生物学的快速发展,基因治疗使得CF这种单基因遗传病的治疗有了重大突破[31]。目前CF基因治疗主要针对CFFR功能缺陷。Ivacaftor(依伐卡托)是2012年首次获批的一种CFTR基因增强剂,可延长CFTR通道开放时间,加速细胞表面氯化物的转运,主要用于≥6岁存在特定突变G551D的CF患者,是首个直接针对病因治疗CF的新型药物[32]。研究显示,Ivacaftor在肺功能改善、体质量增加、减少肺部病变急性加重及降低汗液Cl-水平方面均有显著效果,2周后病情即可得到改善并能维持48周,同时未见严重不良反应。也有研究报道,CF患者停用Ivacaftor后肺功能出现严重受损[33]。Orkambi (鲁玛卡托-依伐卡托)于2015年获批用于治疗12岁以上存在F508del突变的CF患者[34]。目前针对其他突变的特异性基因治疗研究也正在进行,开启了治疗CF的新篇章。
除及时诊断及治疗,对此类患儿的长期管理及密切随访也至关重要。对CF患儿应长期进行营养监测,包括体质量指数等指标监测,并及时进行营养指导。CF患儿均应按时接受指定的免疫接种,尤其是流感疫苗。CF患儿在随访过程中,应及时对严重感染进行评估与治疗,积极的抗生素治疗是患者获得良好预后的关键[35,36]。此外,对CF患者心理疏导也至关重要,CF患儿及其看护者的心理问题,特别是焦虑和抑郁,可影响治疗依从性及远期预后,因此,一旦发现,应接受恰当的心理治疗。
近20多年来,CF患者的生存期显著提高,这在很大程度上得益于传统治疗技术的改进及治疗方法的创新,但对建立一个完善的中国CF患者的诊疗体系,仍是急需解决的临床问题。

























