
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
先天性粒细胞减少症(CN)是一种罕见的先天性骨髓衰竭性疾病,以基因缺陷所致慢性粒细胞减少为特征,伴或不伴造血及免疫系统外的异常;包括先天性重症粒细胞减少症(SCN)和周期性粒细胞减少症(CyN)2种类型,国外报道CN患病率为0.7/100万~1.0/100万[1],编码中性粒细胞弹性蛋白酶(NE)的ELANE基因突变是最常见的基因缺陷[1]。为提高对该罕见病的认识,总结南京医科大学附属儿童医院收治1例伴SH2D1A基因突变的ELANE基因突变相关的综合征性SCN。现报道如下。
患儿,男,6岁4个月,因"反复发热、咳嗽、口腔溃疡、鹅口疮6年"于2017年2月入院。患儿于4月龄时开始出现发热、咳嗽,中性粒细胞绝对计数(ANC)<0.5×109/L;随后每年至少患"呼吸道感染"10余次、"重症肺炎"1次,并反复出现口腔溃疡、鹅口疮,且出牙即开始龋齿;每次查ANC均显著低于正常。多次体液免疫及细胞免疫检查均未见异常。入院前1个月,患儿再次出现发热、进行性咳嗽加重,有黄痰,胸片示双肺炎症。外院予"比阿培南、阿奇霉素、伏立康唑"等治疗,病情无改善。患儿第2胎第2产,足月顺产,出生体质量3.7 kg,否认宫内窘迫及出生后窒息史,因病未按计划添加辅食及预防接种;父母及9岁姐姐均体健。
入院查体:神志清,精神欠佳,生长发育落后,营养不良貌;双眼微凸、下颌前突,面色苍白,口唇无发绀,双眼睑、阴囊、双下肢凹陷性水肿,口腔黏膜见广泛不易拭去白斑及散在溃疡,20枚黑色龋齿,咽充血,双侧扁桃体Ⅱ度;呼吸急促,轻度三凹症,双肺闻及广泛中细湿啰音;心音有力,腹膨软,腹壁静脉显露,有轻度压痛,无反跳痛,肝右肋下2.5 cm,质软,脾未触及,移动性浊音阳性;杵状指(趾)。辅助检查:肌内注射粒细胞集落刺激因子(G-CSF)前:10次ANC (0.21~0.46)×109/L (正常范围2×109/L~7×109/L)、单核细胞(0.82~2.94)×109/L (正常范围0.12×109/L~0.80×109/L);CD3 56.49% (59.00%~84.00%)、CD4+ 27.99% (31.00%~60.00%)、CD8+ 30.06% (13.00%~38.00%)、CD4+/CD8+ 0.93 (0.90~3.60)、B淋巴细胞(BLC)40.27% (7.00%~22.00%);IgA 7.63 g/L (0.58~1.00 g/L)、IgM 1.1 g/L (1.1~1.8 g/L)、IgG 37.70 g/L (6.60~10.39 g/L);自身抗体全套及染色体核型等均未见异常;入院时清蛋白19 g/L;5次痰培养大肠埃希菌生长、1次白色念珠菌生长;Epstein-Barr病毒(EBV)衣壳抗原-IgA、IgG及核抗原IgG均阳性,血EBV-DNA 6.47×106 copies/L。G试验997.3 ng/L (>100.0 ng/L)。骨髓细胞形态学:粒系增生减低,中晚幼粒细胞、杆状核及分叶核明显减低,单核细胞增生活跃。影像学:(1)双肺炎症,右上肺脓肿,胸腹腔积液;(2)迷走左椎动脉、奇静脉扩张,主动脉旁侧支血管迂曲增粗,下腔静脉肝段缺如,左肺静脉未显示(图1)。电子支气管镜:右上支气管黏膜内血管增粗凸出、搏动(图2)。武汉康圣达医学检验所使用安捷伦外显子芯片捕获+高通量测序:(1)ELANE基因(c.641G>A,p.G214E)(图3A),父母均无此突变(图3B、图3C)。(2) SH2D1A基因(c.7G>T,p.A3S)(图3D),父亲无此突变(图3E)。遗传自母亲(图3F)。未发现其他基因异常。诊断:伴SH2D1A基因突变的ELANE基因突变相关的综合征性SCN、重症肺炎。治疗:予保护性隔离,先后予"美罗培南、利奈唑胺、伏立康唑、复方磺胺甲 ?唑(SMZco)"抗感染,"红细胞、清蛋白、丙种球蛋白"等支持治疗,G-CSF 5 μg/(kg·d)升白细胞,3~10 d后症状、体征逐渐改善,4次查ANC(0.65~2.04)×109/L,3周后胸腹腔积液吸收、肺炎好转出院,后继续予利奈唑胺等序贯治疗3周后,目前正进行SMZco口服预防治疗、定期随访,等待造血干细胞移植(HSCT)。嘱监护人注意患儿的保护性隔离。所有检查及治疗患儿监护人均知情同意并签署知情同意书,并经医院医学伦理委员会批准(批准文号:201702002-1)。
?唑(SMZco)"抗感染,"红细胞、清蛋白、丙种球蛋白"等支持治疗,G-CSF 5 μg/(kg·d)升白细胞,3~10 d后症状、体征逐渐改善,4次查ANC(0.65~2.04)×109/L,3周后胸腹腔积液吸收、肺炎好转出院,后继续予利奈唑胺等序贯治疗3周后,目前正进行SMZco口服预防治疗、定期随访,等待造血干细胞移植(HSCT)。嘱监护人注意患儿的保护性隔离。所有检查及治疗患儿监护人均知情同意并签署知情同意书,并经医院医学伦理委员会批准(批准文号:201702002-1)。
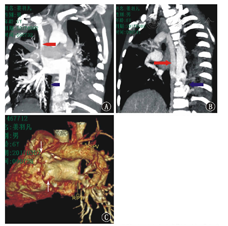
Cardiography of child with severe congenital neutropenia caused by ELANE and SH2D1A mutation
注:MIP:最大密度投影;A:冠状位MIP图,红箭头示上腔静脉蓝箭头示下腔静脉未见;B:矢状位MIP图,红箭头示扩张奇静脉,蓝箭头示迂曲增粗侧支血管;C :VR重建图,左侧肺动静脉细小,右侧肺动静脉形态正常MIP:maximum intensity projection;A:coronal MIP map, the red arrow shows the superior vena cava, the blue arrow shows no inferior vena cava;B: sagittal MIP reconfiguration, the red arrow shows dilated azygos vein, the blue arrow shows tortuous dilated collateral vessels;C: virtual reality reconstruction images, the arrows shows the left pulmonary arteriovenous is small, the right pulmonary arteriovenous is normal
Cardiography of child with severe congenital neutropenia caused by ELANE and SH2D1A mutation
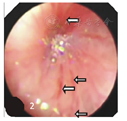
The arrows showing the blood vessels in the bronchial mucosa were obviously enlarged and protruded in child with severe congenital neutropenia caused by ELANE and SH2D1A mutation
The arrows showing the blood vessels in the bronchial mucosa were obviously enlarged and protruded in child with severe congenital neutropenia caused by ELANE and SH2D1A mutation
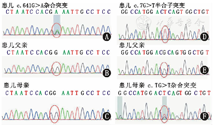
Sanger sequencing results of ELANE and SH2D1A gene of patient with severe congenital neutropenia caused by ELANE and SH2D1A mutation and his parents
注:A~C:ELANE基因;D~F:SH2D1A基因 A-C:ELANE gene;D-F:SH2D1A gene
Sanger sequencing results of ELANE and SH2D1A gene of patient with severe congenital neutropenia caused by ELANE and SH2D1A mutation and his parents
作为一种临床表型和基因型均异质性的疾病,CN临床表现:(1)仅累及血液、免疫系统的非综合征性CN,或并皮肤、心脏等病变的综合征性CN;(2)ANC周期性减少的CyN及持续重度减少(<0.5×109/L)的SCN,甚至ANC不减少,仅引起中性粒细胞胞外杀菌陷阱(NETs)缺陷[2];(3)遗传方式包括常染色体显性或隐性遗传、X-连锁隐性遗传及散发等[3]。目前认为,50%~60%的CN为位于19P13.31、包含5个外显子的ELANE基因突变所致,使得ANC及其杀菌功能同时降低[1];致病机制以未折叠蛋白反应(UPR)、NETs为主[2,4]。因Wnt信号可增加内质网对UPR的耐受性,ELANE基因突变并不影响单核细胞的分化和功能[5],本例患儿骨髓中单核细胞即增生活跃。
本例患儿4月龄起即发现ANC持续重度减少,反复、多部位、多病原感染,中性粒细胞成熟停滞于早幼粒细胞阶段,染色体核型未见异常,亦未发现体液、细胞免疫缺陷及其相关基因缺陷,排除体液、细胞及联合免疫缺陷病,符合SCN诊断标准[1]。同时排除感染性、医源性、血液或自身免疫性疾病等继发性CN因素。HGMDpro数据库报道c.641G>A,p.G214E基因突变可致病。但本例患儿同时伴ELANE和SH2D1A基因突变。遗传自母亲、基因序列位于Xq25的SH2D1A基因突变(c.7G>T,p.A3S),致其编码的信号淋巴细胞活化分子相关蛋白(SAP)降低,引起CD4+/CD8+倒置,记忆BLC减少;SH2D1A基因突变者在感染EBV后BLC和CD8+过度增殖、活化并广泛器官浸润,可能引起暴发性传染性单核细胞增多症、噬血细胞性淋巴组织增生综合征等,病死率极高[6]。本例患儿存在EBV感染,CD4+/CD8+倒置、BLC增多,但无典型X-连锁淋巴组织增生性疾病(XLP)表现,不除外与ELANE基因突变致SCN降低了炎症反应有关,与文献报道的1例ELANE基因突变并其他基因突变患者病情减轻一致[7]。本例患儿虽小于6月龄即疑诊SCN,但直到学龄期才确诊ELANE基因突变,应与该病罕见、合并症多(龋齿、发音模糊、杵状指趾及体态、面容、血管畸形等)、经验不足、缺乏诊断条件等有关。
SCN的治疗关键在于控制感染,常需多种抗生素联合静脉用药。本例患儿采用美罗培南、利奈唑胺等抗球菌及杆菌的抗生素,且因G试验明显增高予伏立康唑抗真菌感染治疗,并予"红细胞、清蛋白、丙种球蛋白"等支持治疗,肌内注射G-CSF以升高ANC,出院后SMZco预防治疗,随访感染控制良好,但考虑到SCN患者恶性肿瘤和严重感染的终生危险,HSCT作为唯一能同时治愈ELANE和SH2D1A基因异常的疗法已被列为选项[1,6]。
综上,对于生命早期出现反复多部位感染、ANC持续低下者,应行骨髓检查及基因序列分析。应用G-CSF可升高ANC但不能完全纠正功能缺陷[2],需注意保护性隔离,反复感染者可考虑预防效益高于风险的SMZco等预防治疗,每年行骨髓细胞遗传学分析,防止进展为急性髓系白血病(AML)/骨髓增生异常综合征(MDS)甚至死亡等严重不良事件[8],把握HSCT指征。

























