
探讨1例ACADVL基因突变导致极长链酰基辅酶A脱氢酶缺乏症(VLCADD)的临床特点及基因突变类型,总结导致VLCADD的基因突变类型。
对内蒙古医科大学附属医院儿科重症监护病房2016年8月收治的1例VLCADD患儿的临床表现及治疗过程进行分析,经患儿监护人同意并签署知情同意书后,行基因(患儿及父母)二代高通量测序分析加PCR测序验证,并以"ACADVL"为检索词,查阅检索中国知网(CNKI)数据库、万方数据库(建库至2016年12月)、PubMed数据库(建库至2016年12月)、在线人类孟德尔遗传数据库(OMIM)及HGMD数据库相关文献,对所发现的突变位点进行分析并总结。
依据患儿主诉、查体及相关检查化验,提示患儿患有VLCADD,给予口服左卡尼汀及中链脂肪酸饮食1周后复查代谢筛查恢复正常。随访3个月后患儿肝脏明显回缩,为右肋下3 cm,剑突下1 cm可触及。基因检测发现ACADVL(NM_000018.3) Exon7:c.608C>T;p.(Pro203Leu)(杂合)和ACADVL(NM_000018.3) Exon18:c.1748C>T;p.(Ser583Leu)(杂合),为复合杂合突变,致病基因分别来自父母双方。文献检索提示迄今有73种突变类型与VLCADD有关,结合本例患儿的突变类型共有75种突变类型。
本例VLCADD临床表现特殊,兼有心律失常、肝大和低血糖表现,经过系统治疗可部分控制疾病的发展。基因检测可以明确诊断,上述2种突变可能为VLCADD的新发突变。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
极长链酰基辅酶A脱氢酶缺乏症(VLCADD)是一种常染色体隐性遗传性线粒体脂肪酸β氧化代谢障碍疾病,于1993年Bertrand等[1]首次报道。根据其临床表现的轻重分为3个亚型,即严重型、中间型和肌病型[2]。因VLCADD缺乏特异性的临床表现及实验室检查方法,故其诊治过程及鉴别诊断较为困难。现对内蒙古医科大学附属医院2016年8月收治的1例VLCADD患儿的临床表现、诊断思路和治疗经过进行报道,并结合其相关文献分析该病的临床特点,以期加深临床医师对此病的认识。
患儿,男,11个月,因"咳嗽14 d,喘息10 d,发热伴嗜睡2 d"入院。第1胎,第1产,无围生期窒息史,生长发育史正常,家族史无异常。
患儿体质量11 kg,身长78 cm,头围45 cm,体温37.5 ℃,脉搏160次/min,呼吸40次/min,血压97/54 mmHg(1 mmHg=0.133 kPa),精神差,嗜睡,急性病容,咽充血,双肺可闻及固定细湿啰音及喘鸣音。心率160次/min,心音低钝,呈奔马律,腹部膨隆。腹围56 cm,无压痛及反跳痛,肝脏右肋下约3 cm,剑突下1 cm,质地稍硬,无触痛,脾未及,移动性浊音阴性,余查体未见异常。
心电图:窦性心动过速。胸部及腹部CT:(1)支气管肺炎;(2)脂肪肝;(3)胆胰脾未见异常。血糖:0.3 mmol/L,酮体:+++,血常规:白细胞(WBC):38.56×109/L,红细胞(RBC):4.14×1012/L,血红蛋白(Hb):92 g/L,血小板(PLT):772×109/L,余项未见异常。C反应蛋白(CRP):16.63 mg/L;PCT:2.17 μg/L;血生化:肌酶及血脂正常,ALT:53 U/L,AST:107 U/L,二氧化碳(CO2)结合力:17.4 mmol/L,总蛋白:50 g/L,清蛋白:36 g/L,球蛋白:14 g/L,肌钙蛋白T:0.033 μg/L。脑钠肽前体:1 358 ng/L;血浆乳酸:1.73 mmol/L;铜蓝蛋白:0.19 g/L;血氨:51.3 μmol/L。血串联质谱结果(2016-9-1):肉豆蔻烯酰肉碱(C14:1)及肉豆蔻二烯酰肉碱(C14:2)增高,分别为0.69和0.25,伴游离肉碱(C0,4.8)及多种酰基肉碱[丙酰肉碱(C3),0.28、丁酰肉碱(C4),0.04]降低,提示极长链酰基辅酶A脱氢酶缺乏症。尿液有机酸气相色谱分析:无显著异常;治疗1周后复查:与前次结果比较,增高的酰基肉碱降至正常。分子遗传检测报告见表1、图1。

ACADVL基因(NM_000018.3)分子遗传检测报告
Molecular genetic testing report of ACADVL gene (NM_000018.3)
ACADVL基因(NM_000018.3)分子遗传检测报告
Molecular genetic testing report of ACADVL gene (NM_000018.3)
| 核苷酸变化 | 氨基酸变化 | 变异类型 | 家系来源验证情况 | |
|---|---|---|---|---|
| 受检者之父 | 受检者之母 | |||
| c.608C>T | p.(Pro203Leu) | 杂合 | 未发现变异 | 杂合变异 |
| c.1748C>T | p.(Ser583Leu) | 杂合 | 杂合变异 | 未发现变异 |

诊断:(1)VLCADD;(2)重症肺炎。给予患儿肺炎基础治疗,并给予口服左卡尼丁及中链脂肪酸饮食,1周后复查代谢筛查恢复正常。随访3个月后患儿肝明显回缩,为右肋下1 cm,剑突下未触及。监护人签署知情同意书后,对患儿基因行二代高通量测序分析及PCR验证(包括父母),综上所述,根据患儿病史、查体特点及血尿遗传代谢筛查、基因检测结果,患儿确诊为VLCADD。该患儿已接受左卡尼汀及保肝药物治疗8 d,后复测遗传代谢病氨基酸和酰基肉碱谱分析,结果较前明显好转,患儿规律服用药物治疗3个月余后,并食用富含中链脂肪酸的油类及奶粉,肝脏较前明显回缩。
以"ACADVL"为检索词,检索中国知网(CNKI)数据库、万方数据库(建库至2016年12月)、PubMed数据库(建库至2016年12月)、在线人类孟德尔遗传数据库(OMIM)以及HGMD数据库,迄今已报道166种ACADVL基因突变类型,国内仅有5篇[3,4,5,6]文章报道了22例ACADVL基因突变导致的VLCADD。国外文献报道总结诸多ACADVL基因突变导致的VLCADD患者。现将国内外文献报道的73种与VLCADD有关的基因突变类型及本例2个新发突变共75例的资料总结如下,有105-BP DEL等31个是致病突变(表2),其中错义突变有27个,无义突变有4个,缺失有2个,且以错义突变最为常见。且有报道称c.343delG(p.Gln100Valfs)与VLCADD心肌病型有相关性[7],c.637G>A(p.Ala213Thr)与该病的肝型有相关性[8]。有c.-63_-49dupGGGCGTGCAG-GACGC等9个突变是良性变异,NG_007975.1:g.5088T>C等26个突变是临床意义未明,NM_000018.3(ACADVL):c.623-8C>T等7个致病性有争议的。
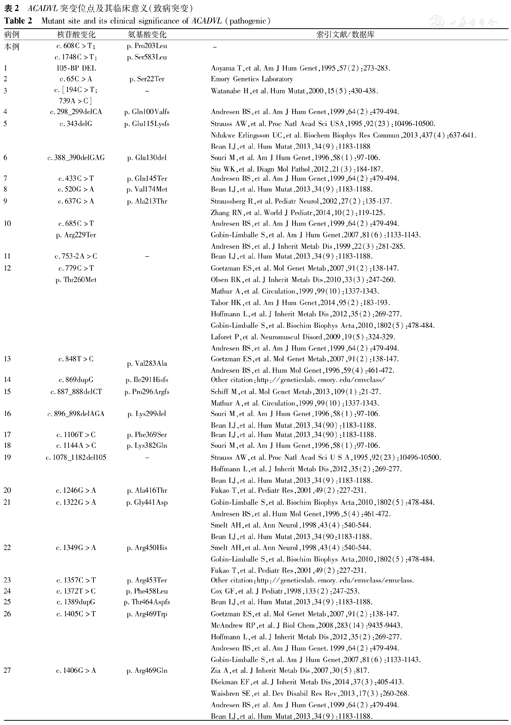
ACADVL突变位点及其临床意义(致病突变)
Mutant site and its clinical significance of ACADVL (pathogenic)
ACADVL突变位点及其临床意义(致病突变)
Mutant site and its clinical significance of ACADVL (pathogenic)
| 病例 | 核苷酸变化 | 氨基酸变化 | 索引文献/数据库 |
|---|---|---|---|
| 本例 | c.608C>T; | p.Pro203Leu | - |
| c.1748C>T; | p.Ser583Leu | ||
| 1 | 105-BP DEL | Aoyama T,et al.Am J Hum Genet,1995,57(2):273-283. | |
| 2 | c.65C>A | p.Ser22Ter | Emory Genetics Laboratory |
| 3 | c.[194C>T;739A>C] | - | Watanabe H,et al.Hum Mutat,2000,15(5):430-438. |
| 4 | c.298_299delCA | p.Gln100Valfs | Andresen BS,et al.Am J Hum Genet,1999,64(2):479-494. |
| 5 | c.343delG | p.Glu115Lysfs | Strauss AW,et al.Proc Natl Acad Sci USA,1995,92(23):10496-10500. |
| Ndukwe Erlingsson UC,et al.Biochem Biophys Res Commun,2013,437(4):637-641. | |||
| Bean LJ,et al.Hum Mutat,2013,34(9):1183-1188 | |||
| 6 | c.388_390delGAG | p.Glu130del | Souri M,et al.Am J Hum Genet,1996,58(1):97-106. |
| Siu WK,et al.Diagn Mol Pathol,2012,21(3):184-187. | |||
| 7 | c.433C>T | p.Gln145Ter | Andresen BS,et al.Am J Hum Genet,1999,64(2):479-494. |
| 8 | c.520G>A | p.Val174Met | Bean LJ,et al.Hum Mutat,2013,34(9):1183-1188. |
| 9 | c.637G>A | p.Ala213Thr | Straussberg R,et al.Pediatr Neurol,2002,27(2):135-137. |
| Zhang RN,et al.World J Pediatr,2014,10(2):119-125. | |||
| 10 | c.685C>T | Andresen BS,et al.Am J Hum Genet,1999,64(2):479-494. | |
| p.Arg229Ter | Gobin-Limballe S,et al.Am J Hum Genet,2007,81(6):1133-1143. | ||
| Andresen BS,et al.J Inherit Metab Dis,1999,22(3):281-285. | |||
| 11 | c.753-2A>C | - | Bean LJ,et al.Hum Mutat,2013,34(9):1183-1188. |
| 12 | c.779C>T | Goetzman ES,et al.Mol Genet Metab,2007,91(2):138-147. | |
| p.Thr260Met | Olsen RK,et al.J Inherit Metab Dis,2010,33(3):247-260. | ||
| Mathur A,et al.Circulation,1999,99(10):1337-1343. | |||
| Tabor HK,et al.Am J Hum Genet,2014,95(2):183-193. | |||
| Hoffmann L,et al.J Inherit Metab Dis,2012,35(2):269-277. | |||
| Gobin-Limballe S,et al.Biochim Biophys Acta,2010,1802(5):478-484. | |||
| Laforet P,et al.Neuromuscul Disord,2009,19(5):324-329. | |||
| Andresen BS,et al.Am J Hum Genet,1999,64(2):479-494. | |||
| 13 | c.848T>C | p.Val283Ala | Goetzman ES,et al.Mol Genet Metab,2007,91(2):138-147. |
| Andresen BS,et al.Hum Mol Genet,1996,59(4):461-472. | |||
| 14 | c.869dupG | p.Ile291Hisfs | Other citation:http://geneticslab.emory.edu/emvclass/ |
| 15 | c.887_888delCT | p.Pro296Argfs | Schiff M,et al.Mol Genet Metab,2013,109(1):21-27. |
| Mathur A,et al.Circulation,1999,99(10):1337-1343. | |||
| 16 | c.896_898delAGA | p.Lys299del | Souri M,et al.Am J Hum Genet,1996,58(1):97-106. |
| Bean LJ,et al.Hum Mutat,2013,34(90):1183-1188. | |||
| 17 | c.1106T>C | p.Phe369Ser | Bean LJ,et al.Hum Mutat,2013,34(90):1183-1188. |
| 18 | c.1144A>C | p.Lys382Gln | Souri M,et al.Am J Hum Genet,1996,58(1):97-106. |
| 19 | c.1078_1182del105 | - | Strauss AW,et al.Proc Natl Acad Sci U S A,1995,92(23):10496-10500. |
| Hoffmann L,et al.J Inherit Metab Dis,2012,35(2):269-277. | |||
| Bean LJ,et al.Hum Mutat,2013,34(9):1183-1188. | |||
| 20 | c.1246G>A | p.Ala416Thr | Fukao T,et al.Pediatr Res,2001,49(2):227-231. |
| 21 | c.1322G>A | p.Gly441Asp | Gobin-Limballe S,et al.Biochim Biophys Acta,2010,1802(5):478-484. |
| Andresen BS,et al.Hum Mol Genet,1996,5(4):461-472. | |||
| Smelt AH,et al.Ann Neurol,1998,43(4):540-544. | |||
| Bean LJ,et al.Hum Mutat,2013,34(90:1183-1188. | |||
| 22 | c.1349G>A | p.Arg450His | Smelt AH,et al.Ann Neurol,1998,43(4):540-544. |
| Gobin-Limballe S,et al.Biochim Biophys Acta,2010,1802(5):478-484. | |||
| Fukao T,et al.Pediatr Res,2001,49(2):227-231. | |||
| 23 | c.1357C>T | p.Arg453Ter | Other citation:http://geneticslab.emory.edu/emvclass/emvclass. |
| 24 | c.1372T>C | p.Phe458Leu | Cox GF,et al.J Pediatr,1998,133(2):247-253. |
| 25 | c.1389dupG | p.Thr464Aspfs | Bean LJ,et al.Hum Mutat,2013,34(9):1183-1188. |
| 26 | c.1405C>T | p.Arg469Trp | Goetzman ES,et al.Mol Genet Metab,2007,91(2):138-147. |
| McAndrew RP,et al.J Biol Chem,2008,283(14):9435-9443. | |||
| Hoffmann L,et al.J Inherit Metab Dis,2012,35(2):269-277. | |||
| Andresen BS,et al.Am J Hum Genet.1999,64(2):479-494. | |||
| Gobin-Limballe S,et al.Am J Hum Genet,2007,81(6):1133-1143. | |||
| 27 | c.1406G>A | p.Arg469Gln | Zia A,et al.J Inherit Metab Dis,2007,30(5):817. |
| Diekman EF,et al.J Inherit Metab Dis,2014,37(3):405-413. | |||
| Waisbren SE,et al.Dev Disabil Res Rev,2013,17(3):260-268. | |||
| Andresen BS,et al.Am J Hum Genet,1999,64(2):479-494. | |||
| Bean LJ,et al.Hum Mutat,2013,34(9):1183-1188. |
| 病例 | 核苷酸变化 | 氨基酸变化 | 索引文献/数据库 |
|---|---|---|---|
| 28 | c.1504C>G | p.Leu502Val | Other citation:http://geneticslab.emory.edu/emvclass/emvclass. |
| 29 | c.1532+1G>A | p.Arg511Gln | Other citation:http://geneticslab.emory.edu/emvclass/emvclass. |
| 30 | c.1679-6G>A | — | Schiff M,et al.Mol Genet Metab,2013,109(1):21-27. |
| Andresen BS,et al.Hum Mol Genet,1996,5(4):461-472. | |||
| Andresen BS,et al.Am J Hum Genet,1999,64(2):479-494. | |||
| Roe CR,et al.J Clin Invest,2002,110(2):259-269. | |||
| Cox GF,et al.J Pediatr,1998,133(2):247-253. | |||
| 31 | c.1837C>T | p.Arg613Trp | Goetzman ES,et al.Mol Genet Metab,2007,91(2):138-147. |
| Strauss AW,et al.Proc Natl Acad Sci USA,1995,92(23):10496-10500. | |||
| Souri M,et al.Am J Hum Genet,1996,58(1):97-106. | |||
| Aoyama T,et al.Am J Hum Genet,1995,57(2):273-283. | |||
| Laforet P,et al.Neuromuscul Disord,2009,19(5):324-329. | |||
| Hale DE,et al.Pediatr Res,1005,19(7):666-671. | |||
| Gobin-Limballe S,et al.Am J Hum Genet,2007,81(6):1133-1143. | |||
| Souri M,et al.Am J Hum Genet,1996,58(1):97-106. |
注:该表所有突变位点来源于人类孟德尔遗传数据库(OMIM)及PubMed数据库(建库至2016年12月) All mutation sites in this table are derived from OMIM and PubMed database(built up from the beginning to December 2016)
自1993年由Bertrand等[1]首次报道VLCADD以来,其临床特点相对清晰。该病是由于细胞线粒体内脂肪酸β氧化中的关键酶极长链酰基辅酶A脱氢酶(VLCAD)基因先天缺陷所致的一种罕见的常染色体隐性遗传性疾病。该病共有3种类型[3],临床上以心肌病型最为常见,主要见于新生儿及婴儿早期发病者,病情危重,病死率极高,常伴心肌受累,且有多脏器衰竭,如肥厚型心肌病、扩张型心肌病、心包积液、心律失常、肌张力降低、瑞氏综合征、反复低血糖发作及肝大。肝型主要多见于儿童期,临床表现为低血糖和异常低血酮体征、肝大等。肌病型主要见于青少年期至成人期,为迟发型,临床症状较轻微,多表现为反复发作性横纹肌溶解,伴肌肉疼痛性痉挛性肌肉酸痛,较少发生低血糖等上述症状。本例患儿主要表现为肝大、低血糖,考虑诊断为肝型。心律失常可能是由于肺部感染诱发,但也可能是属于兼有肝型和心肌病型的混合型,需要密切随访以后是否会再次出现心律失常等心肌病的表现。
VLCADD的诊断需依赖于临床表现、实验室检查及血液液相串联质谱、基因检测等手段[9],实验室血液相关检查可有血清心肌酶谱水平升高,肌红蛋白尿,尿常规和肾功的异常。血液液相串联质谱检测[10]可发现多种长链酰基肉碱谱水平增高,其中C12和C14明显升高,以C14:1升高最为明显[11],同时还有C2明显降低,依此来鉴别VLCADD的不同临床类型,且常以C14:1作为VLCADD的识别标记。故在疾病初诊时,当患儿伴有继发性肉碱缺乏时,不能仅根据C14:1升高的程度来判断疾病的严重程度[5]。本例患儿血液遗传代谢筛查显示C14:1及C14:2增高,伴游离肉碱C0及多种酰基肉碱降低,符合该病的诊断。对于尿液进行有机酸分析,有时可见VLCADD患儿酮体水平低下及二羧酸尿症,本例患儿并未见异常。
VLCADD是由ACADVL突变所致,ACADVL位于染色体17p13.1,长约5.4 kb,其中包含20个外显子,编码655个氨基酸前体蛋白,位于线粒体内膜,属于脂酰辅酶A脱氢酶(ACAD)家族成员之一。ACADVL的突变谱高度异质[7],且该基因的基因型与VLCADD的表现型之间存在明显相关性[12]。无义突变多见于临床表现危重的患儿,无义突变导致酶活性的完全丧失,从而引发患儿的心肌病变、肝脏损害及难以纠正的代谢紊乱;错义突变多见于婴幼儿时期起病的患儿,且多见于肝型和肌病型中,因该突变基因的酶活性尚未全部丧失,而且婴幼儿极少进行长时间过度的体力活动,故临床表现及症状较为轻微[13,14]。本例患儿发生的突变为错义突变,正符合该型的特点。与VLCADD相关的ACADVL基因突变类型多样,包括缺失、重复、无义等多种突变类型,根据表2得知致病突变类型有31种,其中突变类型中错义突变有27个,无义突变有4个,缺失有2个等等,其中以错义突变最为常见。Zhang等[8]学者对患有VLCADD的7例中国患者进行研究,研究表明每例患者均带有不同的基因突变,并将基因突变类型报道如下:p.S22X,p.W427X,p.A213T,p.G222R,p.R450H,c.296-297delCA,c.1605+1G>T,p.S72F,p.Q100X,p.M437T,p.D466Y,且明确表明该基因的基因型与该疾病的临床表现有相关性。虽然ACADVL高度异质,但Miller等[15]报道p.V283A为一种较为常见的错义突变位点。本例患儿基因检测结果:ACADVL(NM_000018.3) Exon7:c.608C>T;p.(Pro203Leu)(杂合)和ACADVL(NM_000018.3) Exon18:c.1748C>T;p.(Ser583Leu)(杂合),为复合杂合突变。父亲:ACADVL(NM_000018.3) Exon18:c.1748C>T;p.(Ser583Leu)(杂合),母亲ACADVL(NM_000018.3) Exon7:c.608C>T;p.(Pro203Leu)(杂合)。其中c.608C>T(编码区第608号核苷酸由C变为T)、c.1748C>T(编码区第1748号核苷酸由C变为T)的复合杂合核苷酸变异,上述变异分别导致第203号氨基酸由Pro变为Leu(p.Pro203Leu)、第583号氨基酸由Ser变为Leu(p.Ser583Leu),均为错义变异,上述变异均可能导致蛋白质功能受到影响。患儿上述变异分别遗传自父母,其父母均只有携带其中1个杂合变异,为复合杂合突变。目前,该变异尚未被dbSNP138、ESP6500、HGMD和千人基因组数据库等基因数据库收录,不属于核苷酸多态性。变异c.608C>T和c.1748C>T对于其他疾病的致病性已经有文献报道:c.608C>T的变异与人类琥珀酸半醛脱氢酶缺乏症(Succinic Semialdehyde Dehydrogenase,SSADH)[16]和遗传性视网膜劈裂症[17]相关。c.1748C>T的变异与儿童起病有关的神经系统疾病[18]相关。故这2种突变位点已在在其他疾病中报道提示该突变位点有致病性。对于该类遗传方式,复合杂合变异可能导致发病。在患儿ACADVL基因所发现的复合杂合变异分别遗传自患儿的父母,父母均为杂合子,符合常染色体隐性遗传方式,故以上变异很可能是导致患儿发病的致病性变异。此患儿为外显子7和18区的复合杂合突变,致病基因分别来自父母双方,与上述检索结果对照,考虑本例患儿的突变可能为ACADVL的新发突变类型。
VLCADD的治疗主要包括:(1)合理饮食:以高碳水化合物及低脂饮食为主,多补充中链三酰甘油,减少长链脂肪酸的摄入。(2)补充左旋肉碱:可明显缓解患儿异常的心功能,减少空腹低血糖的发生率。(3)避免空腹等[19]。本例患儿经过上述治疗,获得较为满意的疗效,更加说明本例的诊断是正确的,上述2个突变位点可能为ACADVL的新发突变类型,但本研究的不足之处是缺乏功能相关分析其是否为致病突变基因,需要进一步研究并密切随访,以提高生存质量。
总之,通过本例患儿的诊疗过程,不仅扩宽了VLCADD的基因谱,而且还加强了对此类较为罕见的遗传代谢性疾病的了解及认识,提高了临床医师诊断该疾病的水平,为基因治疗和遗传咨询奠定了基础。

























