探讨Currarino综合征患者的临床表现及运动神经元与胰腺同源框基因(motor neuron and pancreas homeobox 1 gene,MNX1基因)突变的特点。
利用染色体芯片技术对Currarino综合征患者进行全基因组水平微缺失/微重复检测,并复习文献比较相似基因型Currarino综合征患者的临床表型。
纳入2例Currarino综合征患者。例1,女,7 d,因"反复呕吐"就诊,入院查体发现患者面容特殊,足月小样儿,视不追物,腹部胀气严重;患儿肛门狭窄,肠道造影提示中肠旋转不良;心脏彩超提示卵圆孔未闭或房间隔缺损;头颅超声提示颅内出血,骶尾椎磁共振成像(MRI)示脊髓栓系,骶尾椎发育畸形;染色体芯片技术检测发现7号染色体q36.1q36.3区域缺失1个拷贝(7.89 Mb),14q32.33区域重复1个拷贝(2.20 Mb);患儿父母该区域未见异常。例2,女,1岁3个月,因"整体发育落后"就诊;患儿面容特殊,右眼上睑下垂,语言、运动均落后,肛门狭窄,MRI示隐性脊柱裂;染色体芯片技术检测发现7号染色体q35q36.3区域缺失1个拷贝(15.00 Mb)。文献复习:MNX1基因所在的染色体区域缺失导致的Currarino综合征存在特殊面容、智力障碍及生长落后的特征,国内未见该类型的报道。
在国内首次报道了2例因染色体微缺失导致的Currarino综合征,丰富了临床医师对该病的认识。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Currarino综合征是由先天性肛门直肠畸形、骶前肿块以及骶骨畸形组成的三联征,最早由放射科医师Currarino报道了3例典型病例[1,2]。研究发现,位于染色体7q36区域的运动神经元与胰腺同源框基因(motor neuron and pancreas homeobox 1 gene,MNX1基因)的突变或单倍剂量不足与该病的发生发展相关[3,4],不同的基因型可导致不同的临床表型,且表型差异较大[5]。因为Currarino综合征的临床表现多变,所以该病仅依靠临床表型诊断较为困难。本研究结合华中科技大学同济医学院附属武汉儿童医院诊断的2例Currarino综合征,探讨该病的临床表现及基因突变的特点,以加深临床医师对该病的认识。
例1,女,7 d,因"反复呕吐"就诊。入院查体:体温36.5 ℃,面容特殊(图1A),足月小样儿,神志清楚,反应可,皮肤颜色红润,弹性正常;双肺呼吸音清,未闻及干湿性啰音;心率144次/min,心音有力,心脏各听诊区未闻及杂音;四肢未见异常,活动无受限,视不追物,腹部外形膨隆,以上腹部为主,胃型无,肠型无,全腹柔软(图1B);肝肋下1 cm可触及,质软。神经系统查体:肌张力正常,吸吮反射、双手握持反射以及拥抱反射均减弱。进一步查体发现患儿肛门狭窄,肠道造影提示中肠旋转不良;疑似先天性巨结肠;心脏彩超提示卵圆孔未闭或房间隔缺损;头颅超声提示颅内出血,骶尾椎磁共振成像(MRI)示脊髓栓系,骶尾椎发育畸形,见图2。患儿父母身体健康,非近亲结婚,且双方家庭均无类似疾病家族史。患儿母亲34岁,孕40周,自然分娩;患儿系第4胎,第2产,患儿有一哥哥,表型正常;其余2胎为死胎或死产。
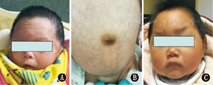
注:A:例1面部特点;B:例1腹部特点;C:例2特殊面部特征 A:facial features of patient 1;B:severe abdominal distention in patient 1;C:patient 2 showed a special face
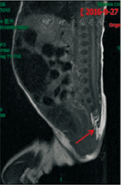
例2,女,1岁3个月,因"发育迟缓"就诊。入院查体:体温36.8 ℃,神志清醒,面容异常(图1C);皮肤色泽正常,反应可;双肺呼吸运动对称,呼吸音听诊正常,未闻及啰音;心脏各听诊区未闻及杂音;四肢未见异常,大动作及精细动作均落后同龄儿童,Gesell发育量表显示,适应性59,大运动46,精细动作53,语言59,个人社交58。患儿先天性右眼上睑下垂,眼神交流差。家长口述:患儿出生15 d右眼睁开,胀气严重,排便困难。神经系统检查:肌张力未见明显异常,生理反射正常,Babinski征阴性,Brudzinkski征阴性。进一步查体发现患儿肛门狭窄;外院脊椎MRI示隐性脊椎裂的症状(图2)。头颅MRI未见异常。患儿系第1胎,第1产,足月自然分娩,父母身体健康,非近亲结婚,且双方家庭均无类似疾病家族史。
本研究得到患儿监护人知情同意,并得到武汉儿童医院医学伦理委员会审核通过(审批号:武儿医2017021)。
经过患儿监护人知情同意后行单核苷酸多态性(single-nucleotide polymorphism,SNP)芯片检测。乙二胺四乙酸(EDTA)抗凝管取患儿及其父母外周静脉血2 mL,利用OMEGA公司(美国佐治亚州)的全血基因组DNA提取试剂盒提取DNA,利用超微量分光光度计SMA-4000(美林恒通,北京)测定浓度,调整DNA质量浓度为50 mg/L。采用Illumina OmniZhonghua-8v芯片进行染色体微缺失/微重复检测,芯片产生的原始数据采用KaryoStudio软件进行分析。
根据芯片分析的结果,选择异常区域内的MNX1基因进行荧光定量分析。MNX1基因上游引物序列:5′-GTGCAGGGAGACCTGGAACAG-3′;下游引物序列:5′-ACCCTGGGTGACACAGCAAGA-3′。选择单拷贝基因36B4为内参基因,上游引物序列:5′-CAGCAAGTGGGAAGGTGTAATCC-3′;下游引物序列:5′-CCCATTCTATCATCAA-CGGGTACAA-3′。利用SYBR荧光染料,在ABI7500荧光定量PCR仪器上以36B4基因为参考进行MNX1基因的相对定量检测,检测结果利用仪器自带软件进行相对定量分析。
利用中国期刊全文数据库以及美国国立生物技术信息中心(NCBI)数据库查阅Currarino综合征相关的文献,并比较分析相似基因型的Currarino综合征患者的临床表型及差异。
例1存在2处异常:7号染色体q36.1q36.3区域存在大小为7.89 Mb的缺失,该区域包括MNX1、SHH、WD重复蛋白60基因(WDR60)、二肽基肽酶6基因(DPP6)基因等23个在线人类孟德尔遗传(OMIM)基因;14q32.33区域重复1个拷贝,大小为2.20 Mb,该区域包括丝氨酸/苏氨酸蛋白激酶1基因(AKT1)、RNA聚合酶Ⅲ转录起始因子亚基基因(BRF1)等15个OMIM基因。例2提示7号染色体q35q36.3区域缺失1个拷贝,大小为15.00 Mb,该区域包括MNX1、SHH、KCNH2、CNTNAP2等62个OMIM基因,见图3。
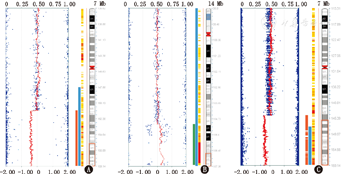
注:A:例1患儿7号染色体部分缺失;B:例1患儿14号染色体存在微重复;C:例2患儿7号染色体部分缺失 A:the deletion of 7q36.1q36.3 region in patient 1;B:the duplication of 14q32.33 region in patient 2;C:the deletion of 7q35q36.3 region in patient 2
利用荧光定量PCR验证SNP芯片的结果,结果显示,相对于健康人群,2例患儿MNX1基因均缺失1个拷贝(患者该区域为1个拷贝),而患儿父母该区域未见异常。
检索中国期刊全文数据库,万方数据库以及PubMed数据库(截止到2017年6月30日),国内有文献报道Currarino综合征,多数患者以肛门直肠畸形、骶骨畸形以及骶骨肿块的临床表型诊断,有少数患者发现MNX1基因携带点突变;国内未见含MNX1基因区域大片段缺失导致Currarino综合征的报道。且国内报道的Currarino综合征均未合并智力障碍、生长发育迟缓以及特殊面容等临床表现。
国外报道了多例MNX1基因异常导致的Currarino综合征,其中约15例为7号染色体q36区域(包括MNX1基因)缺失。比较分析发现:大片段缺失的患者几乎均有生长发育迟缓、智力低下及特殊面容的临床表现,而MNX1基因点突变的Currarino综合征患者未见以上临床表现。此外存在大片段缺失的患者合并其他临床表现(如泌尿系统畸形、先天性巨结肠等)的比例较高。
Currarino综合征通常表现为骶前区肿块、骶骨畸形及先天性肛门直肠畸形的三联征,是一种与MNX1基因异常相关的常染色体显性遗传病[6]。其临床表型多变,部分患者除以上三联征外还存在多系统异常,如智力障碍、整体发育迟缓、面容特殊、泌尿系统畸形等[7]。MNX1基因位于7号染色体q36区域,全长5 801 bp,含4外显子,编码含有401个氨基酸的蛋白;该基因在胰腺的正常发育过程及脊髓中运动神经元的分化过程发挥至关重要的作用[8]。研究发现,在几乎所有的家族性Currarino综合征患者及约30%的散发病例中可检测到MNX1基因异常[9]。
本研究例1患儿存在肛门狭窄、脊髓栓系以及骶尾椎发育畸形等典型Currarino综合征的临床表现,还存在肠扭转及先天性巨结肠的表型,因其年龄较小,其整体发育情况未做评估。全基因组芯片的结果提示,例1患儿7号染色体q36.1q36.3区域缺失7.89 Mb,包括导致Currarino综合征的MNX1基因;此外研究发现该区域的SHH基因异常可导致先天性巨结肠及高度近视等症状[10],因此,推测例1先天性巨结肠的症状与该基因的杂合缺失有关,但文献有报道少数MNX1基因点突变所致Currarino综合征患者存在先天性巨结肠[11]。例1染色体14q32.33区域重复2.20 Mb,该区域不存在剂量敏感的基因[5],推测该区域的重复可能和患儿的临床表型无关。
本研究例2患儿存在发育落后、肛门狭窄、隐性脊椎裂等症状,且一直存在腹部胀气的症状。全基因组芯片的结果提示例2患儿7号染色体q35q36.3区域缺失15.00 Mb,缺失的区域包括MNX1、SHH、KCNH2、CNTNAP2及EZH2等OMIM基因。KCNH2基因的单倍剂量不足可导致遗传性长QT综合征[12],且有文献报道包含KCNH2基因缺失的Currarino综合征患者婴儿期存在心律不齐的症状[5],本例患儿未行心电图检测。研究发现,CNTNAP2基因异常与自闭症相关[13],例2患者存在语言落后、眼神交流稍差的表型。EZH2基因异常可导致Weaver综合征,该综合征临床表现为运动发育落后,肌张力高,智力障碍、对称的身材高大等症状[14];本例患儿应长期随访,监测是否有以上并发症的发生。
文献复习发现,国内未见大片段缺失导致Currarino综合征的报道;携带MNX1基因内的突变(包括错义突变、无义突变、小片段的缺失/重复等)的患者临床上仅表现为Currarino综合征典型的特征,而患者不表现为生长发育迟缓及智力障碍,极少数患者存在特殊的面容[5,15]。7号染色体q36区域(包括MNX1基因)缺失的患者除典型的临床表型外,还存在智力低下,发育迟缓,特殊面容的表型,此外部分患者还合并泌尿系统畸形,先天性巨结肠,前脑无裂畸形等[5,16];本研究中的2例患者均为缺失突变,临床特点与文献报道的一致。因此,推测单独的MNX1基因突变可能不会导致发育迟缓,7号染色体q36区域的基因缺失可能导致基因连续缺失综合征即Currarino综合征合并发育迟缓及其他畸形。不同基因型的患者存在不同的临床表型,因此临床医师可依据患者的临床表现选择MNX1基因直接测序技术或者选择染色体芯片技术检测。
综上所述,本研究首次在国内报道2例由7号染色体q36区域缺失导致的Currarino综合征,加深国内临床医师对该疾病的认识,此类型的患者除典型的Currarino综合征的临床表型外,还合并智力障碍、生长发育落后以及特殊面容,染色体芯片技术可协助诊断。


























