
回顾性分析郑州大学第三附属医院儿童康复科收治的1例婴儿期White-Sutton综合征(WSS)并甲状腺功能减低症患儿的临床特征、实验室检查结果、病历资料和基因检测结果,结果显示本例患儿在婴儿期即出现运动、智力发育明显落后,行为异常,肌张力低下,听觉传导通路异常,胃肠道问题则表现为经常性腹胀、便秘,但本例患儿食欲尚可,体质量为10 kg;头面部特征表现为头扁平、眼距宽、四肢短粗。实验室检查显示甲状腺功能减低。基因检测发现POGZ:NM_015100:exon19:c.C2590T:p.R864X。提示临床表现为精神运动发育迟缓、头面部异常、肌张力低下、自闭倾向、胃肠道功能紊乱、甲状腺功能减低症的患儿应考虑WSS的可能,应进一步检查以明确诊断。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
White-Sutton综合征(WSS)是一种罕见的神经发育异常遗传病,常造成精神运动发育迟缓、视听觉损伤、胃肠道功能障碍等问题。目前国内有关WSS的文献报道极少,现对2017年12月郑州大学第三附属医院儿童康复科收治的1例合并甲状腺功能减低症的WSS患儿的临床特征进行总结报道,以提高儿科医师对本病的认识水平。
患儿,女,10个月18 d,以"10个月18 d竖头不稳"为代主诉入院。患儿系第1胎,第1产,足月自然分娩,出生体质量3.9 kg,出生第2天因"新生儿肺炎、缺氧缺血性脑病(HIE)"于当地医院治疗15 d好转出院,新生儿期存在自发动作少、吃奶呛咳情况。既往有"肺炎"病史,父母均体健,否认近亲结婚,否认有家族遗传病史及类似脑性瘫痪(脑瘫)病史。体格检查:神志清,精神欠佳,表情淡漠,营养状况可,皮肤白皙,发黄稀疏,头面部发育不良,头围40 cm,头扁平、眼距宽、鼻孔上翻;四肢短小,腹部膨隆,叩诊呈鼓音。反应迟钝,不知寻找声源,难于逗笑,自主发声少,头控制欠佳,拉起时头后垂,不知主动抓物、手支撑及翻身不能完成,坐位全前倾,四肢肌力3级,肌张力低,围巾征阳性,内收肌角180°,腘窝角180°,足背屈角60°,握持反射、拥抱反射消失,踝阵挛(-),腱反射未引出。头颅磁共振成像(MRI):白质髓鞘化发育落后于4月龄儿,双侧额颞部蛛网膜下腔增宽;心脏彩超:卵圆孔未闭;脑干听觉诱发电位:双侧90 db Ⅰ波潜伏期延长,高频听阈70 db;视觉诱发电位:双侧视觉通路未见异常;脑电图:正常脑电图;双下肢肌电图:未见明显异常。甲状腺功能五项:促甲状腺激素(TSH)2.2 mIU/L,三碘甲状原氨酸(T3) 1.96 pmol/L,甲状腺素(T4) 59.19 pmol/L↓,游离三碘甲状原氨酸(FT3) 5.21 pmol/L,游离甲状腺素(FT4) 9.87 pmol/L↓;血氨8.7 μmol/L;同型半胱氨酸3.8 μmol/L;神经元特异性烯醇化酶19.86 μg/L;染色体核型分析:46,XX,染色体检查所示条带未见明显异常;染色体微阵列检测报告:未检出具有临床意义的基因拷贝数改变和大片段纯合子;血代谢筛查:尿胍基乙酸略高。征得监护人同意后行遗传病基因检测,本研究通过医院医学伦理委员会批准[批准文号:(2017)医伦审第(09)号]。检测报告:先证者发现可以解释或部分解释临床表型的一个致病变异,可能为新发变异;染色体坐标位置chr1:151378921,变异位置、碱基及氨基酸改变信息POGZ:NM_015100:exon19:c.C2590T:p.R864X(图1)。父母:并未携带该变异(图1),但不排除生殖细胞嵌合的可能。治疗经过:入院完善相关检查检验后给予患儿口服甲状腺素片、功能训练、推拿治疗、电子生物反馈等综合康复治疗1个月,患儿竖头较前稳定,可独坐片刻,双手可主动抓物,病情好转出院。
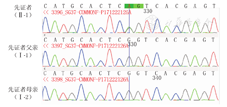
WSS属于常染色体显性遗传病,临床表现为婴儿期即出现精神运动发育迟缓及特殊的面部特征。其他特征还包括肌张力减退、感觉神经听力受损、视力缺陷、关节松弛、胃肠道问题,相当数量的患儿会出现自闭症。致病基因是位于染色体1q21.3的POGZ基因,大部分为新发变异致病,编码锌指蛋白[1]。POGZ基因调控有丝分裂,在人体内集中表达于婴儿或成人脑组织,在分子网络中对神经功能发挥至关重要的调控作用[2]。当异常的POGZ蛋白在脑组织神经元内大量出现时,很有可能通过改变细胞周期影响神经元细胞的增殖而成为引起疾病的原因[3]。本例患儿父母均未患病,未携带相关基因,因此患儿考虑为新发变异。
WSS患者个体间的临床表现差异大,有些患者小时候喂食困难,但长大后身材会过度肥胖,也会有胃酸反流与便秘的情况。其他还有低血糖导致的癫痫、过动、睡眠障碍、行为与精神方面异常,有时会有自残的行为。虽然大部分患者有自闭症的倾向,但有些患者则相反,喜欢社交且态度友善[4]。总结国外的33例患者发现,多数患者表现为发育迟缓、智力障碍、行为障碍、偏头痛、自闭行为,诊断年龄均在1岁以上,其中男19例,女14例,男性略多于女性。但33例均未合并甲状腺功能减低症[4,5,6,7,8]。
甲状腺功能减低症是儿科常见内分泌疾病之一,甲状腺素可促进物质氧化、氧化磷酸化作用加强,促进新陈代谢;同时还可促进蛋白质、糖、脂肪、水盐代谢,可促进大脑、生长发育。甲状腺功能低下可见特殊面容、智力低下,记忆力、注意力下降,运动发育障碍,生长发育迟缓及消化道功能紊乱[9]。本例患儿出生后苯丙酮尿症、先天性甲状腺功能减低症筛查未见异常,入院后发现甲状腺功能减低症。因为临床患者少,既往研究资料并未涉及到甲状腺功能的研究,所以甲状腺功能减低症是否是WSS的一个继发性改变并不清楚。本例患儿合并甲状腺功能减退症,在婴儿期即出现运动、智力发育明显落后,行为异常,肌张力低下,听觉传导通路异常,胃肠道问题则表现为经常性腹胀,便秘,但本例患儿食欲尚可,体质量为10 kg;头面部特征表现为头扁平,眼距宽,四肢短粗。
在WSS的治疗方面,由于本病属于罕见病,临床病例少,治疗经验不足,目前尚无特效治疗方法,主要采用对症治疗。本例患儿经1个月康复治疗,患儿头部控制能力较前提高,可独坐片刻;但患儿智力及行为表现方面未见明显改善。由于患儿治疗时间短,随访时间不长,对其临床预后尚不清楚。
综上,在临床上表现为精神运动发育迟缓、头面部异常、肌张力低下、自闭倾向、胃肠道功能紊乱、甲状腺功能减低症的患儿应考虑WSS的可能,并进一步检查以明确诊断。
所有作者均声明不存在利益冲突

























