
分析1例丙酮酸脱氢酶E1α缺乏症患儿PDHA1基因的变异特点及其相应的临床症状,为临床诊断、遗传咨询和产前诊断提供依据。
对1例丙酮酸脱氢酶E1α缺乏症患儿的临床症状、生化检查及治疗效果进行分析;运用高通量测序技术对患儿及其父母外周血进行基因组DNA检测,运用Sanger测序对患儿姐姐进行阳性位点验证。
患儿,男,3个月,持续性高乳酸血症,血乳酸9.05~11.04 mmol/L,不会抬头,反应欠灵敏。头颅磁共振成像提示双侧侧脑室略显僵直,胼胝体体部变薄,双侧枕骨发育不对称。3 h脑电图提示睡眠期左侧前中颞区为主尖波、尖慢复合波散发;睡眠期广泛性中-高波幅2.0~3.0 Hz慢波阵发。高通量基因测序结果提示,患儿PDHA1基因c.1243_1257dup15(p.V415-N419dup)半合子变异,结合临床表现确诊为丙酮酸脱氢酶E1α缺乏症。患儿母亲及姐姐未携带该变异,且排除母亲致病位点嵌合体的可能性。最后患儿因治疗无效于1岁3个月死亡。
PDHA1基因c.1243-1257dup15重复变异为新发变异,该变异可导致丙酮酸脱氢酶E1α缺乏症;丙酮酸脱氢酶E1α缺乏症的确诊需进行分子诊断。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
线粒体丙酮酸脱氢酶复合物(pyruvate dehydrogenase complex,PDHC)是由6种酶组成的位于线粒体基质的复合物,其催化丙酮酸不可逆的氧化脱羧转化成乙酰辅酶A,将糖类有氧氧化与三羧酸循环和氧化磷酸化连接起来,在细胞线粒体呼吸链代谢中的作用至关重要[1]。丙酮酸脱氢酶复合物缺乏症(pyruvate dehydrogenase complex deficiency,PHD)是导致线粒体能量代谢障碍最常见的遗传代谢病之一,同时也是婴儿及儿童原发性高乳酸血症和早发性退行性神经变性病的常见病因。PHD多以酸中毒和神经系统症状为主,临床常表现为致命的高乳酸血症、脑结构异常、精神运动发育迟缓、间歇性运动失调及癫痫等[2,3,4]。PHD患者多数因PDHA1基因变异引起E1α亚基功能缺陷而发病,即丙酮酸脱氢酶E1α缺乏症。PDHA1基因是X染色体连锁遗传,男性发病率较高,但由于X染色体随机失活,女性也可发病。现分析1例丙酮酸脱氢酶缺乏症男童的临床资料及基因检测结果,为该症的临床诊断、遗传咨询和产前诊断提供依据。
患儿(先证者),男,3个月,系第3胎,第2产;第1胎女,现5岁,发育同健康同龄儿;第2胎于胎龄3个月自然流产。患儿胎龄35周剖宫产出生(胎盘前置),出生体质量1.5 kg,出生后因早产、低出生体质量、新生儿窒息(轻度)、新生儿呼吸窘迫综合征等于2016年7月进入东莞市妇幼保健院新生儿科治疗41 d。3个月时因俯卧位不能抬头、视听及逗引反应欠灵敏于2016年10月首次入东莞市妇幼保健院康复科。
发育评估:3个月时粗大运动功能测量(gross motor function measure,GMFM):2.35%,程度:较差;发育商(developmental quotient,DQ)和发育年龄(developmental age,DA):DA=1.4个月,DQ=45分,属异常,其中大动作DQ:0分,精细动作DQ:65分,适应能力DQ:32分,语言DQ:65分,社交行为DQ:65分。8个月时GMFM:5.4%,程度:稍差;DA=2.5个月,DQ=33分,属异常,其中大动作DQ:13分,精细动作DQ:33分,适应能力DQ:26分,语言DQ:53分,社交行为DQ:39分。10个月时DA=2.2个月,DQ=23分,属异常,其中大动作DQ:0分,精细动作DQ:26分,适应能力DQ:21分,语言DQ:42分,社交行为DQ:26分。5个月时身高较健康儿童矮8 cm。1岁时不能竖头,不会翻身,四肢肌力肌张力偏低,发育相当于2个月婴儿水平。
头颅磁共振成像(MRI)检测及脑电图检查:3个月时患儿头颅MRI显示双侧侧脑室略显僵直,胼胝体体部变薄,患儿脑实质髓鞘化纠正胎龄约相当于3个月水平;双侧枕骨发育不对称。3 h脑电图显示为异常婴幼儿脑电图:睡眠期左侧前中颞区为主尖波、尖慢复合波散发,睡眠期广泛性中-高波幅2.0~3.0 Hz慢波阵发。
生化检查:持续性高乳酸血症,血乳酸9.05~11.04 mmol/L。多次生化检查显示血氨正常,心功能及甲状腺功能也均正常。诊断为丙酮酸脱氢酶缺乏症,予生酮饮食,辅酶Q10,左旋肉碱和三维B片(包括B1、B6和B12)治疗,并继续康复治疗。治疗3个月后,血乳酸2.78 mmol/L,恢复正常水平;血生化检查结果显示,血钾(K+) 6.49 mmol/L,浓度偏高考虑为生酮饮食中服用枸橼酸钾预防结石所致,建议减少钾用量。
患儿1岁3个月因支气管炎住院,随后发热,咳嗽加剧,治疗无效死亡。
取得患儿父母知情同意后,抽取患儿及其父母和姐姐外周静脉血各2 mL,乙二胺四乙酸二钾抗凝管保存。送至广州嘉检医学检测有限公司进行遗传性乳酸酸中毒基因包检测,该基因包含71个相关基因。
基因组DNA提取采用"盐析法血液DNA提取试剂盒"(D3312-01,广州美技生物科技有限公司);运用二代测序技术对基因组DNA进行检测(测序仪Nextseq500,illumina);运用二代测序技术对基因阳性位点进行高精度检测以验证母亲是否为致病变异位点的嵌合体,并运用Sanger测序对患儿姐姐进行阳性位点验证。
本研究通过东莞市妇幼保健院医学伦理委员会批准[伦审批第(58)号],患儿监护人均知情同意并签署知情同意书。
应用预测软件PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/)对变异的保守性、致病性及危害性进行预测。检索人类基因组变异数据库(Human Gene Mutation Database,HGMD)、PubMed、Clinvar(https://www.ncbi.nlm.nih.gov/clinvar/)和人类孟德尔遗传在线(OMIM,http://omim.org/)等数据库参考变异相关文献,分析文献内容,参考美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)变异分类指南对其进行分类。
对患儿及其父母进行遗传性乳酸性酸中毒相关的71个基因的高通量测序和分析,发现患儿PDHA1基因发生1个半合子变异c.1243_1257dup15(p.V415_N419dup),其变异为15个氨基酸的整码插入变异(图1),导致第415-419位氨基酸残基重复;父母PDHA1基因未见该变异。母亲外周血基因组DNA的c.1243_1257dup15位点高精度检测(测序深度14062×)结果显示其杂合比例为0;Sanger测序结果提示患儿姐姐未携带该位点变异(图1)。
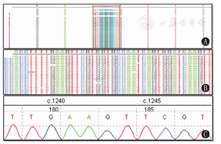
患儿PDHA1基因发生的c.1243_1257dup15位点变异在HGMD和Clinvar数据库中的人群携带频率极低,表明该变异不是人群多态性位点。在PubMed和OMIM等数据库的临床案例中,未见该变异位点的报道。运用预测软件PolyPhen-2对c.1243_1257dup15位点的保守性、致病性及危害性进行预测,结果显示该变异所在的区域是E1α亚基蛋白质的重要组成部分,该区域在人类、黑猩猩、大鼠、恒河猴、猪等不同物种间的氨基酸序列高度保守,且预测该区域影响蛋白质结构和功能的可能性较大。结合以上分析及患儿的临床表现,且依据美国ACMG变异分类指南的标准,c.1243_1257dup15变异被归类为Ⅱ类——可能致病。结合患儿临床表现及基因检测结果,确诊为丙酮酸脱氢酶E1α缺乏症,PDHA1基因c.1243_1257dup15变异可能是其致病原因。
丙酮酸脱氢酶E1α缺乏症是丙酮酸脱氢酶复合物缺乏症最常见的类型,该症在不同年龄阶段有不同的临床表现。新生儿期发病的患者主要表现为严重的乳酸酸中毒和脑部发育不全。婴儿期发病的患者多表现为发育迟缓、肌张力异常、共济失调、脑性瘫痪等进行性神经系统病变,并伴基底核和脑干损害,多在婴幼儿期发展为Leigh综合征[5]。此外,还有少数患者起病时症状较轻,在给予高碳水化合物饮食后出现间歇性共济失调,病情缓慢进展,数年后逐渐发展为Leigh综合征。丙酮酸脱氢酶E1α缺乏症是X连锁遗传疾病,男性发病率高于女性,并且临床表现差异显著;男性发病时间早且症状更严重[6]。由于X染色体随机失活,女性也可发病,但临床表现更为复杂,常见多发畸形如小头畸形、鼻上翻和宽鼻梁等,并乳酸酸中毒的女性患者多预后不良,常在新生儿期至婴儿期死亡;但也有一些轻型女性患者终身不发病[7]。本例患儿男性,新生儿期发病,主要表现为持续性的严重乳酸酸中毒和脑胼胝体体部变薄,双侧枕骨发育不对称,且精神运动发育迟缓,是典型的新生儿期发病症状。
丙酮酸脱氢酶E1α缺乏症患者多数由PDHA1基因变异引发。PDHA1基因编码E1α亚基,全长约1.5 bp,包含11个外显子,共编码390个氨基酸。目前,国外至少报道了300余例经PDHA1基因确诊的丙酮酸脱氢酶E1α缺乏症患者,已知的PDHA1基因致病变异多达160余种,包括错义变异、无义变异、剪接变异、插入/缺失变异等,其中常见的是错义变异和插入/缺失变异[8,9]。插入/缺失变异在丙酮酸脱氢酶E1α缺乏症患者的发生率约为20%,这种变异常导致碱基序列发生移码而引起氨基酸翻译提前终止。目前,关于中国人群PDHA1基因变异的报道很少,国内仅见报道2个已知错义变异和一个新发的插入变异[10,11]。本研究发现的变异c.1243_1257dup15为插入变异,发生在第11外显子,是中国人群在PDHA1基因上发现的第2例插入变异,且该变异为新发变异,在患儿母亲外周血中未检测到嵌合体的存在,亦尚未见有文献报道。本例c.1243_1257dup15变异的报道丰富了中国人群PDHA1基因变异数据库。
本例患儿新生儿期发病,多次住院治疗,但一直未明确病因,于11个月时行基因检测,结合临床表现和基因检测结果,诊断为丙酮酸脱氢酶E1α缺乏症,给予生酮饮食、辅酶Q10、左旋肉碱和三维B片治疗后状况略有好转。随后患儿因支气管炎而发热、咳嗽治疗无效于1岁3个月死亡。新生儿期发病的丙酮酸脱氢酶E1α缺乏症患儿预后较差,多在婴儿或儿童期死亡,但早期给予生酮饮食、硫辛酸、左旋肉碱及辅酶Q10等可以改善部分临床症状和预后,因此,早诊断、早治疗是该症治疗的关键[12]。因丙酮酸脱氢酶E1α缺乏症患儿临床表现缺乏特异性,诊断困难,所以基因检测对该症的确诊起重要作用。此外,对患儿家系进行PDHA1基因变异检测,可以发现无症状或症状轻微的致病基因携带者,也为遗传咨询和产前诊断提供依据。
所有作者均声明不存在利益冲突

























