
探讨SCN1A基因突变的Dravet综合征(DS)患儿不同突变特点与临床表型的相关性及药物疗效。
收集2014年1月至2018年5月在郑州大学第三附属医院小儿神经内科确诊为DS患儿的临床资料,采集患儿及其父母外周血,提取DNA,采用二代高通量基因测序对患儿行癫痫基因包检测,对其父母采用Sanger测序进行家系验证,基因突变阴性患儿采用多重连接探针扩增技术(MLPA)检测SCN1A基因大片段变异,根据年龄对患儿采用Gesell量表或韦氏儿童智力量表(C-WISC)行智力评估。
共收集50例DS患儿,其中38例SCN1A基因突变阳性,突变率为76.0%(38/50例),以错义突变[50.0%(19/38例)]、移码突变[28.9%(11/38例)]为主。50例患儿平均起病年龄为6.00月龄;68.0%(34/50例)的患儿首次发作以发热(>37.5 ℃)诱发,60.0%(30/50例)的患儿有热水浴惊厥发作史;74.0%(37/50例)的患儿有惊厥持续状态;80.0%(40/50例)的患儿有丛集样发作;92.0%(46/50例)的患儿有≥2种发作类型;30.0%(15/50例)轻度智力落后,38.0%(19/50例)中度智力落后,14.0%(7/50例)重度智力落后;24.0%(12/50例)的患儿1岁前即出现脑电图发作间期异常,脑电图异常的平均年龄为30.12个月;药物有效率排名前3位的为托吡酯[70.0%(28/40例)]、丙戊酸钠[48.0%(24/50例)]、氯硝西泮或氯巴占[45.7%(16/35例)];截断突变组患儿肌阵挛及不典型失神发作出现的时间均早于错义突变组(14.75个月比21.20个月,16.82个月比26.00个月),差异均有统计学意义(均P<0.05);截断突变组患儿丛集样发作比例高于错义突变组[94.7%(18/19例)比63.2%(12/19例)],差异有统计学意义(P<0.05);起病年龄、发作类型、惊厥持续状态比例、智力落后程度、1岁前脑电图异常比例及发作频率与SCN1A基因突变方式及突变位点差异无统计学意义(均P>0.05)。
DS患儿SCN1A基因突变率高,SCN1A基因突变特点与DS临床表型有一定相关性;托吡酯为DS患儿常用药物中最有效的药物。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Dravet综合征(DS)是一种难治性癫痫综合征,国外报道本病发病率为1/20 000~1/40 000[1];其临床发作形式多样,易出现惊厥持续状态,大部分患儿对抗癫痫药物治疗效果差,至成年仍有抽搐发作,并有不同程度智力损害[2]。DS主要是由编码电压门控钠离子通道的α亚基的SCN1A基因突变所致。SCN1A基因突变形式及突变位点多样,目前已超过1 200种SCN1A突变被识别,其中超过900种SCN1A突变类型被报道与DS相关[3],但目前关于这些突变特点与DS患儿临床表型相关性方面的研究较少。本研究分析在郑州大学第三附属医院确诊为DS患儿的临床资料及其SCN1A基因突变特点,并总结其可能存在的关联性,为临床提供参考。
选取2014年1月至2018年5月在郑州大学第三附属医院小儿神经内科就诊的癫痫患儿中临床症状符合DS的50例患儿纳为研究对象。DS的诊断标准[4]:(1)1岁以内起病,首次发作多表现为热性惊厥;(2)1~4岁出现多种形式的无热惊厥,包括全面性或半侧阵挛或强直阵挛发作、肌阵挛发作、不典型失神、局灶性发作等;(3)易出现惊厥持续状态;(4)发作具有热敏性及丛集性特点;(5)早期发育正常,发病后逐渐出现智力及运动发育落后及倒退,可出现共济失调和锥体束征;(6)脑电图早期常无异常,随后可表现为背景差,出现广泛性棘慢波、多棘慢波或局灶性、多灶性癫痫样放电;(7)对抗癫痫药物疗效差。本研究通过郑州大学第三附属医院医学伦理委员会批准[批准文号:(2018)医伦审第43号],患儿监护人均知情同意,并签署知情同意书。
采集建立临床资料登记表(包括姓名、性别、出生日期、围生期情况、发育情况、起病年龄、发作类型及频率、不典型失神及肌阵挛发作出现时间、用药史及疗效、重要辅助检查、家族史等),通过与患者家属面谈或电话询问的方式,尽可能多地追溯病史资料并详细记录。
在家属知情并签署知情同意书后采集患儿及家系成员的外周血各2 mL,置于含乙二胺四乙酸(EDTA)抗凝试管中,外送至基因检测公司进行癫痫基因包检测,对患儿采用二代高通量基因测序,对其父母采用Sanger测序进行家系验证,并通过人类基因突变数据库及其他数据库进行数据分析,明确致病性、可疑致病性基因突变;基因突变阴性的患儿应用多重连接探针扩增技术(MLPA)进行SCN1A基因大片段变异检测。
应用SPSS 21.0统计软件进行统计分析。计量资料用 ±s表示,数据在满足正态性和方差齐性的条件下,采用t检验;计数资料比较采用χ2检验或Fisher′s确切概率法,有序定性资料采用秩和检验,P<0.05为差异有统计学意义。
±s表示,数据在满足正态性和方差齐性的条件下,采用t检验;计数资料比较采用χ2检验或Fisher′s确切概率法,有序定性资料采用秩和检验,P<0.05为差异有统计学意义。
50例DS患儿平均起病年龄为6个月,68.0%(34/50例)的患儿首次发作以发热(>37.5 ℃)诱发;32%(16/50例)的患儿首次发病为无热惊厥,随后再表现为热敏感性;60.0%(30/50例)的患儿病程中有过热水澡惊厥发作史;74.0%(37/50例)的患儿有惊厥持续状态(30 min~3.5 h);80.0%(40/50例)的患儿有丛集样发作(24 h内≥2次抽搐);92.0%(46/50例)的患儿有≥2种发作类型,其中强直阵挛发作94.0%(47/50例),肌阵挛发作70.0%(35/50例),不典型失神发作56.0%(28/50例),局灶性发作74.0%(37/50例),全面性阵挛发作44.0%(22/50例),强直发作10.0%(5/10例)。1岁内平均发作频率为1.34次/月,1~5岁平均发作频率为1.89次/月,5岁以后(11例)发作频率较前减少,其中3例患儿发作频率10~15次/年,2例患儿发作频率2~3次/年,其余患儿发作频率4~5次/年。
50例患儿中18.0%(9/50例)智力发育正常(患儿年龄13.04~36.02个月,平均年龄25.18个月),30.0%(15/50例)轻度智力落后,38.0%(19/50例)中度智力落后,14.0%(7/50例)重度智力落后。
约62.0%(31/50例)的患儿出现脑电图发作间期异常,24.0%(12/50例)的患儿1岁前即出现脑电图发作间期异常;异常脑电图表现为背景慢化、局灶性或广泛性癫痫波发放等;出现脑电图异常的平均年龄为30.12个月。
50例患儿中,38例SCN1A突变阳性,突变率为76.0%(38/50例),其中错义突变50.0%(19/38例)、移码突变28.9%(11/38例)、无义突变7.9%(3/38例),剪切突变7.9%(3/38例),大片段缺失5.3%(2/38例);错义突变中,73.6%(14/19例)位于编码核心区(S4-S6区),26.3%(5/19例)位于其他编码区,其中5例中3例患儿具有相同的基因突变位点(c.1880G>A,p.Arg627Lys,结构域DⅠ~DⅡ);97.4%(37/38例)为新生突变,1例突变来源于母亲;1例GABRA1突变阳性,1例PCDH19突变阳性,10例患儿基因检测均阴性。
患儿均口服2种及2种以上药物进行治疗,最多者可达8种药物,药物有效性(发作减少>50%)回顾性分析提示,丙戊酸钠48.0%(24/50例)有效,托吡酯70.0%(28/40例)有效,加托吡酯后最长可控制1.5年无发作,氯硝西泮或氯巴占45.7%(16/35例)有效,左乙拉西坦29.7%(11/37例)有效,奥卡西平或卡马西平76.2%(16/21例)加重,19.0%(4/21例)无加重亦无改善,4.8%(1/21例,该例患儿为PCDH19基因突变)有效;拉莫三嗪80.0%(4/5例)加重,20.0%(1/5例)疗效不确切,苯巴比妥11.1%(1/9例)有效,11.1%(1/9例)加重,余无疗效或不确切。
将研究对象按SCN1A基因突变方式分为错义突变组(19例)和截断突变组(19例)(移码突变、无义突变、剪切突变、大片段缺失),2组患儿的末次随访年龄比较差异无统计学意义,见表1。截断突变组患儿肌阵挛及不典型失神发作出现的时间均早于错义突变组(14.75个月比21.20个月;16.82个月比26.00个月),差异均有统计学意义(均P<0.05),见表1。截断突变组患儿丛集样发作比例高于错义突变组(94.7%比63.2%),差异有统计学意义(P<0.05),见表1。2组患儿的起病年龄、惊厥持续状态发作比例、发作类型、智力落后程度、1岁前的发作频率及脑电图发作间期的异常比例差异均无统计学意义,见表1。
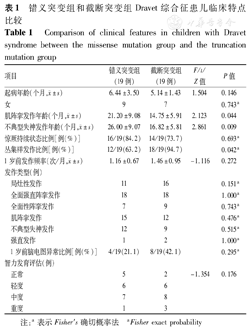
错义突变组和截断突变组Dravet综合征患儿临床特点比较
Comparison of clinical features in children with Dravet syndrome between the missense mutation group and the truncation mutation group
错义突变组和截断突变组Dravet综合征患儿临床特点比较
Comparison of clinical features in children with Dravet syndrome between the missense mutation group and the truncation mutation group
| 项目 | 错义突变组(19例) | 截断突变组(19例) | F/t/Z值 | P值 | |
|---|---|---|---|---|---|
起病年龄(个月, ±s) ±s) | 6.44±3.50 | 5.14±1.43 | 1.504 | 0.146 | |
| 女 | 9 | 7 | 0.743a | ||
肌阵挛发作年龄(个月, ±s) ±s) | 21.20±9.08 | 14.75±5.91 | 2.123 | 0.044 | |
不典型失神发作年龄(个月, ±s) ±s) | 26.00±9.07 | 16.82±5.81 | 2.861 | 0.009 | |
| 惊厥持续状态比例[例(%)] | 16/19(84.2) | 14/19(73.7) | 0.693a | ||
| 丛集样发作比例[例(%)] | 12/19(63.2) | 18/19(94.7) | 0.042a | ||
1岁前发作频率(次/月, ±s) ±s) | 1.16±0.67 | 1.46±0.95 | -1.116 | 0.272 | |
| 发作类型(例) | |||||
| 局灶性发作 | 11 | 16 | 0.151a | ||
| 全面强直阵挛发作 | 18 | 18 | 1.000a | ||
| 全面性阵挛发作 | 7 | 9 | 0.743a | ||
| 肌阵挛发作 | 15 | 12 | 0.476a | ||
| 不典型失神发作 | 12 | 9 | 0.515a | ||
| 强直发作 | 1 | 2 | 1.000a | ||
| 1岁前脑电图异常比例[例(%)] | 4/19(21.1) | 8/19(42.1) | 0.295a | ||
| 智力发育评估(例) | |||||
| 正常 | 5 | 2 | -1.354 | 0.176 | |
| 轻度 | 6 | 6 | |||
| 中度 | 7 | 8 | |||
| 重度 | 1 | 3 | |||
注:a表示Fisher′s确切概率法 aFisher exact probability
将研究对象中错义突变患儿按突变位点分为编码核心区(S4-S6)突变组(14例)和非核心区突变组(5例),2组患儿末次随访年龄差异无统计学意义,见表2。2组患儿的起病年龄、发作类型、惊厥持续状态的发作比例、1岁前的发作频率及脑电图发作间期的异常比例、智力落后程度差异均无统计学意义,见表2。
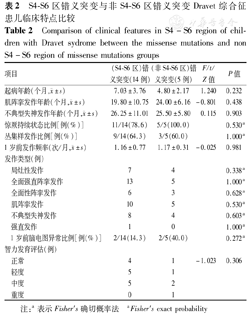
S4-S6区错义突变与非S4-S6区错义突变Dravet综合征患儿临床特点比较
Comparison of clinical features in S4-S6 region of children with Dravet sydrome between the missense mutations and non S4-S6 region of missense mutations groups
S4-S6区错义突变与非S4-S6区错义突变Dravet综合征患儿临床特点比较
Comparison of clinical features in S4-S6 region of children with Dravet sydrome between the missense mutations and non S4-S6 region of missense mutations groups
| 项目 | (S4-S6区)错义突变(14例) | (非S4-S6区)错义突变(5例) | F/t/Z值 | P值 | |
|---|---|---|---|---|---|
起病年龄(个月, ±s) ±s) | 7.03±3.76 | 4.80±2.17 | 1.240 | 0.232 | |
肌阵挛发作年龄(个月, ±s) ±s) | 19.80±10.75 | 24.00±6.16 | -0.801 | 0.438 | |
不典型失神发作年龄(个月, ±s) ±s) | 26.25±11.01 | 25.50±5.80 | 0.115 | 0.903 | |
| 惊厥持续状态比例[例(%)] | 11/14(78.6) | 5/5(100.0) | 0.530a | ||
| 丛集样发作比例[例(%)] | 9/14(64.3) | 3/5(60.0) | 1.000a | ||
1岁前发作频率(次/月, ±s) ±s) | 1.16±0.77 | 1.17±0.31 | -0.025 | 0.981 | |
| 发作类型(例) | |||||
| 局灶性发作 | 7 | 4 | 0.338a | ||
| 全面强直阵挛发作 | 13 | 5 | 1.000a | ||
| 全面性阵挛发作 | 6 | 3 | 0.628a | ||
| 肌阵挛发作 | 10 | 5 | 0.530a | ||
| 不典型失神发作 | 8 | 4 | 0.603a | ||
| 强直发作 | 1 | 0 | 1.000a | ||
| 1岁前脑电图异常比例[例(%)] | 2/14(14.3) | 2/5(40.0) | 0.272a | ||
| 智力发育评估(例) | |||||
| 正常 | 4 | 1 | -1.023 | 0.306 | |
| 轻度 | 5 | 1 | |||
| 中度 | 5 | 2 | |||
| 重度 | 0 | 1 | |||
注:a表示Fisher′s确切概率法 aFisher′s exact probability
DS是一种难治性癫痫性脑病,常有不同程度的智力损害,其临床发作形式多样,具有热敏感性,早期易被误诊为热性惊厥。本研究中50例DS患儿的平均起病年龄为6月龄,其起病年龄明显要早于热性惊厥的高发年龄1.5岁[7];其次DS患儿惊厥发作易表现为惊厥持续状态及丛集样发作,本研究中显示74.0%的患儿病程中出现惊厥持续状态,最长者可达3.5 h,80.0%的患儿有丛集样发作,与既往文献[7]报道基本一致。另外DS患儿热敏感特征明显,部分患儿低热即可出现惊厥发作。Hattori等[8]研究发现58.7%(27/46例)的DS患儿1岁前即有热水浴惊厥发作史,本研究中显示60.0%(30/50例)的患儿病程中有热水浴惊厥发作史,而临床观察中极少发现热性惊厥患儿有热水浴惊厥发作的情况,由此可见该特征可能对早期诊断DS有一定提示意义。DS患儿虽热敏感特征明显,但部分患儿可以以无热起病,并逐渐表现出热敏感性。许小菁等[7]研究发现32.6%(59/181例)的DS患儿1岁内即出现无热惊厥发作,其中10.5%(19/181例)的患儿首次发作即为无热惊厥,本研究中32.0%(16/50例)的DS患儿首次发病为无热起病,随后再表现出热敏感特征。
随着分子遗传学技术的发展,DS发病机制中遗传因素的作用被研究的越来越多,目前被报道的参与DS发病的基因有SCN1A、PCDH19、SCN1B、GABRA1、GABRG2、CHD2、STXBP1等[4,9,10,11]。其中编码电压门控钠离子通道的α亚基的SCN1A基因是其主要的致病基因,据文献报道由SCN1A突变导致DS占70%~80%,其中新生突变占90%~95%,遗传性突变占5%~10%[12]。本研究中50例DS患儿,38例SCN1A突变阳性,突变率为76.0%,新生突变为97.4%,突变方式以错义突变及移码突变为主,这与文献[12]报道基本一致。
SCN1A基因突变方式多样,包括错义、移码、无义、剪切、大片段缺失及重复等突变形式;错义突变导致个别氨基酸改变;而移码突变及无义突变使翻译终止密码子提前,以致蛋白质表达提前终止,剪切突变、大片段的缺失或重复致使碱基重排,导致转录及翻译异常,以致蛋白表达减少或缺如,统称为截断突变[13]。本研究通过比较错义突变组及截断突变组患儿的临床特点,发现截断突变组患儿肌阵挛及不典型失神发作出现的时间均早于错义突变组(14.75个月比21.20个月;16.82个月比26.00个月),这与Zuberi等[14]的研究一致;而Ragona等[15]在研究影响DS患儿智力的临床因素中发现,肌阵挛发作及不典型失神发作出现时间越早,智力落后越严重。
既往研究表明DS患儿中错义突变位点多位于电压感受区(S4)和门孔区(S5-S6)[3]。本研究显示73.7%(14/19例)的错义突变位于核心编码区(S4-S6),而26.3%(5/19例)则位于非核心区,其中5例中有3例患儿具有相同的基因突变位点(c.1880G>A,p.Arg627Lys,结构域DI-DII)。比较2组患儿的临床特点,2组患儿的起病年龄、发作频率、惊厥发作比例、发作类型、智力落后程度等指标差异均无统计学意义,推测钠离子通道除了已认知的核心功能区外,可能还有未发现的功能区域或存在有某些调节基因在其致病中起作用。
DS患儿早期发育基本正常,随着年龄的增长,患儿常并不同程度智力落后,目前已有研究表明一些临床变量与智力落后程度相关,如惊厥持续状态频率高、1岁前脑电图出现异常、无热惊厥出现年龄越早、肌阵挛及不典型失神发作出现的时间越早,智力落后越明显[15,16,17],而关于SCN1A基因突变类型是否与智力落后程度有关尚无定论。日本大样本(285例)回顾性分析比较截断突变组及错义突变组患儿的智力发育情况,结果表明截断突变组患儿智力落后下降的速率较错义突变组快[18]。Bender等[19]通过动物模型研究发现,SCN1A基因突变可独立于癫痫本身直接对智力产生影响,并认为截断突变者相对错义突变者可能预后差。而Ragona等[15]在回顾性分析26例DS患儿智力发育的影响因素时则认为SCN1A基因突变类型与智力落后程度无关。本研究发现2组患儿的智力发育情况差异无统计学意义,因为本研究样本量较小,所以对于该问题有待于进一步研究论证。
DS是药物难治性癫痫,多数患儿对抗癫痫药物反应差,很难达到完全控制,目前丙戊酸钠、司替戊醇、氯巴占联合使用是国外治疗DS的金标准[20,21],中国癫痫指南则推荐将丙戊酸钠、托吡酯和/或氯巴占作为DS一线治疗方案。本研究中所有患儿均口服2种及以上药物治疗,最多者可达8种;药物疗效回顾性分析发现,药物有效率最高为托吡酯(70.0%)。Kroll-Seger等[22]报道托吡酯治疗DS的有效率为77.8%(28/36例),二者基本一致。另有文献报道,将托吡酯与丙戊酸钠及司替戊醇联合使用,疗效可[20]。本研究中26例患儿因早期诊断不明确应用了钠离子通道阻滞剂,如卡马西平、奥卡西平、拉莫三嗪等,但仅1例患儿(PCDH19突变)应用有效,其余为加重或无效。说明钠离子通道阻滞剂主要加重SCN1A基因突变导致的DS,对于其他少见致病基因突变(包括PCDH19基因)导致的DS并不加重。目前国外正在研发的几种新药对治疗DS可能显示出较好的治疗前景,尤其是大麻二酚及芬氟拉明[23],但现处于临床试验阶段,仍需在随机对照试验中进一步评估药物疗效。
所有作者均声明不存在利益冲突

























