
对武汉儿童医院遗传代谢内分泌科诊断的1例多毛软骨发育不良(即Cantú综合征)患儿的临床资料进行回顾性分析。患儿,女,1岁8个月。毛发浓密1年余。查体:前额突出,较窄,耳位低,鼻梁塌,鼻头大,内眦赘皮,嘴唇厚,高颚弓;皮肤色泽偏黑,颜面部、额头、四肢、腰背部、臀部毛发旺盛。脊柱正侧位片示L1-2椎体形态欠规则。基因检测结果显示ABCC9基因存在杂合突变:c.3347G>A(鸟嘌呤>腺嘌呤),导致氨基酸改变p.R1116H(精氨酸>组氨酸),为已报道的致病突变位点。提示Cantú综合征为罕见的常染色体显性遗传病,以多毛、面容异常、心血管及骨骼异常为临床特点,检测ABCC9基因或KCNJ8基因有助于诊断。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
多毛症的原因很多,根据发病年龄分为先天性或获得性。获得性多毛症可能继发于不同原因,如药物不良反应、内分泌失调、感染性疾病、营养不良及卵巢和肾上腺肿瘤等。而先天性多毛症多因基因异常所致,发病早,易并其他系统异常,常以特殊的异常综合征出现。现分析武汉儿童医院遗传代谢内分泌科诊断的1例因ABCC9基因(OMIM 601439)突变所致的多毛软骨发育不良,即Cantú综合征(OMIM 239580)的临床特点和基因检测结果,以提高临床医师对该病的临床表现及基因诊断的认识。
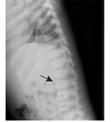
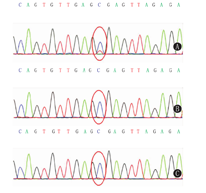
患儿,女,1岁8个月,因"毛发浓密1年余"于2017年10月就诊于武汉儿童医院遗传代谢内分泌科。患儿出生后毛发旺盛,颜色较黑,随年龄增长进行性加重,现多毛主要分布在颜面部、四肢、腰背部、臀部。无吐泻及抽搐,无发热,未行特殊处理。出生史:第2胎,第2产,足月,剖宫产,出生体质量4.55 kg,出生身长53 cm,头围37 cm。发育里程碑:3个月抬头、7个月萌牙、9个月会坐、1.5岁会走路及说话(现能说简单词,如爸爸妈妈、爷爷奶奶等)。身长84 cm(75th~90th)、体质量11 kg(50th~75th),头围51 cm,胸围44 cm,腹围46 cm,上部量55 cm,下部量39 cm。特殊面容,前额突出,较窄,耳位低,鼻梁塌,鼻头大,内眦赘皮,嘴唇厚,高颚弓,掌纹深(图1A、图1B)。皮肤色泽偏黑,皮肤弹性正常,毛发旺盛,主要分布在颜面部、额头、四肢、腰背部、臀部(图1C、图1D)。神志清楚,胸廓较窄,双肺未闻及啰音。心律齐,心音有力,未闻及杂音。腹部膨隆,触诊质软,无压痛及反跳痛,肝脏肋下1.5 cm,脾肋下未触及。四肢肌力及肌张力正常。双乳B1期,无乳头乳晕色素沉着,无阴毛、腋毛生长。父亲身高163.5 cm,母亲身高152.5 cm,遗传靶身高(152±4) cm。血尿便常规、肝肾功能、电解质、心肌酶谱、血脂、乳酸、血氨、骨碱性磷酸酶未见异常。甲状旁腺素33.7 ng/L。25羟维生素D 20.41 μg/L。胰岛素样生长因子-1<25.0 μg/L。生长激素1.15 μg/L。甲状腺功能正常。皮质醇上午8:00时611.05 nmol/L、皮质醇晚20:00时106.91 nmol/L、促皮质素22.8 ng/L。人绒毛膜促性腺激素0.19 IU/L,雌二醇53.67 pmol/L,睾酮0.14 μg/L,促黄体生成素0.01 IU/L,促卵泡生成素5.56 IU/L,泌乳素218.64 μIU/mL,孕酮0.12 μg/L。骨龄1岁6个月。心电图:窦性心动过速。彩超:肝右肋下1.6 cm,脾及双肾未见明显异常。卵圆孔未闭,左心室收缩功能正常。脊柱正侧位片:脊柱各段生理曲度及序列正常,L1-2椎体形态欠规则,椎间隙正常(图2)。四肢及头颅正侧位片:颅盖骨边缘光整,未见明显骨质病变征象。双侧肱骨及尺桡骨未见明显骨质病变,间隙正常。双股骨及胫腓骨未见明显骨质病变,髋膝关节间隙正常。肾上腺CT:未见明显异常。颅脑磁共振成像(MRI)平扫:未见明显异常。Gesell儿童发育量表:患儿一般发育水平约为16个月;患儿适应性、语言发育为边缘水平,其余各项发育正常。抽取患儿及患儿父母外周血2 mL,送北京金准基因科技公司进行全外显子测序。提取外周血白细胞DNA,采用KAPA LTP Library Preparation Kit试剂构建DNA文库。真空浓缩DNA文库样本进行杂交捕获,取捕获后的DNA样本进行高通量测序。所得到的基因变异再使用Sanger测序进行验证。经测序分析显示患儿ABCC9基因cDNA序列的第3 347位的鸟嘌呤突变为腺嘌呤(c.3347G>A),导致氨基酸精氨酸变为组氨酸(p.R1116H)。而父母均未携带(图3)。本研究通过医院医学伦理委员会批准(批准文号:武儿医2017020),患儿监护人知情同意,并签署知情同意书。
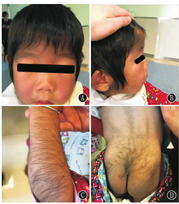
Cantú综合征是多系统受累疾病,极为罕见,中国人群中仅有数例临床报道。本例为极少有明确基因突变的中国Cantú综合征报道。其特征为先天性多毛症(颜面、背部及四肢体毛过多)、粗糙面容(宽阔鼻梁、丰满嘴唇、巨舌、牙龈增生等)、心血管(心室质量增加、动脉导管未闭、心脏扩大、心包积液等,心脏收缩力多正常)和骨骼异常(增厚的颅骨、狭窄的胸腔、宽肋骨、扁平或卵圆形的椎体、脊柱侧凸等,通常无症状性)[1]。出生多为巨大儿伴头围大。智力多正常,但大多数人语言及运动发育迟缓[1]。近年还有少数脑血管扩张屈曲报道[2]。
Cantú综合征为常染色体显性遗传病,迄今已报道50例Cantú综合征中有35例行基因检测,28例存在ABCC9杂合突变,均为错义突变[1,3],2例存在KCNJ8杂合突变[4]。ABCC9和KCNJ8均位于12p12.1,编码ATP敏感性钾通道(KATP)成孔亚基(Kir6.1,KCNJ8编码)和调控亚基(SUR2,ABCC9编码)。其激活突变将降低ATP对通道的抑制作用,使通道持续开放[3,4,5]。ABCC9编码亚基SUR2A(在心脏及骨骼肌中表达)和SUR2B(在平滑肌及毛囊中表达)。本例患儿为ABCC9突变,突变位点c.3347G>A已有报道[3],结合其特征性临床表现,Cantú综合征诊断明确。该综合征基因型及表型相关性仍不明。目前表现为粗糙面容及多毛的有3类疾病:Cantú综合征、肢端肥大症面容综合征及多毛伴肢端肥大症面容综合征[6,7]。临床表现上的交叉表明在分子遗传学层面亦有重叠。ABCC9为明确的Cantú综合征致病基因,同时在另2种综合征中亦检测到该突变[6,8]。目前认为它们均为ABCC9突变所致,Cantú综合征为较严重的表型,余为较温和的表型[6]。
Cantú综合征主要为对症治疗。针对多毛酌情使用脱毛剂。近年亦有报道磺脲类药物可作为潜在的Cantú综合征的治疗手段[5],局部外用磺脲类药物可抑制Cantú综合征引起的毛发过度生长,但仍需大量临床数据证实。对于该病,建议每年监测心电图及心脏彩超以评估心脏大小及功能,同时行脊柱片监测脊柱侧凸情况。若出现明显骨骼及心脏异常,需考虑手术干预。
所有作者均声明不存在利益冲突

























