
对北京儿童医院神经内科诊断的KMT2A基因变异导致的Wiedemann-Steiner综合征1例患儿的临床资料进行回顾性分析,患儿,男,1岁4个月,因"发育迟缓伴多毛"就诊,体质量6.5 kg,身长66 cm,头围42 cm,均小于健康同龄儿童的2个标准差,营养不良,发育落后,不能独走,不会说话,易激惹、好动,肘部及后背皮肤多毛,特殊面容,眉毛浓密、小眼裂、长睫毛、宽眼距、塌鼻梁、球状鼻、薄上唇,肾脏彩超示马蹄肾,右肾盂充盈;头颅磁共振成像示胼胝体压部短,颞极蛛网膜下腔宽,侧脑室后部变尖,枕骨大孔似略窄,全外显子组基因测序发现患儿携带KMT2A基因第27外显子c.7071delC杂合缺失移码变异,导致蛋白质改变为p.S2358Lfs*18,该变异国内外均未见报道。提示对于生长迟缓、发育落后、多毛及特殊面容患者应考虑Wiedemann-Steiner综合征,除考虑进行染色体数目或拷贝数检测外,还需考虑到检测与染色体修饰重构相关的基因。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Wiedemann-Steiner综合征(WSS)是以身材矮小、精神运动发育迟滞、多毛及特殊面容(包括狭窄的眼裂、浓眉毛、长睫毛、薄上唇、塌鼻梁、球状鼻、指趾畸形等)为主要表现的多发畸形综合征[1],因Wiedemann和Steiner分别在1989和2000年首次描述此病而命名[2,3]。Jones等[4]在2012年将WSS的致病基因定位于编码赖氨酸甲基转移酶2A的KMT2A基因,呈常染色体显性遗传。现对北京儿童医院神经内科诊断的KMT2A基因变异导致的1例儿童WSS的临床及基因特点进行报道,旨在提高临床医师对该疾病的认识。
患儿,男,1岁4个月,于2018年4月因"发育迟缓伴多毛"首次就诊于首都医科大学附属北京儿童医院神经内科门诊。患儿系第2胎、第2产,足月因"瘢痕子宫"行剖宫产,出生体质量6.5 kg,身长45.0 cm,头围33.0 cm,Apgar评分1 min、5 min、10 min均为10分。出生后哭声大,四肢姿势正常,否认病理性黄疸,出生后发现眉毛浓密、睫毛长、眼裂小、眼距宽、肘部及后背多毛,当地医院查血常规白细胞23.68×109/L,中性粒细胞0.714,血红蛋白193.0 g/L,C反应蛋白(CRP)13.3 mg/L;心脏彩超示动脉导管未闭、卵圆孔未闭(3月龄时复查正常),无发热、纳差、反应弱、抽搐,考虑"新生儿感染",予抗感染治疗1周后复查血常规+CRP正常。3个月会抬头,4个月会翻身,7个月会独坐,1岁可扶站,1岁2个月可扶走,至今不会爬、不会独走。至今不会说话,可听懂其名。4个月萌牙,现已出齐16颗牙,11个月前囟闭合。智力运动发育无倒退。10月龄时接种卡介苗疫苗,出现局部皮肤红肿及周围皮肤白色丘疹,持续4个月未消退。平时纳食少、畏食、喜甜食,性格易闹、好动。患儿母亲孕期体健,定期产检无异常,第1胎,第1产为女童,现6岁,健康。父母体健,智力均正常,非近期结婚,否认家族史。体格检查:1岁4个月时,体质量6.5 kg,身长66.0 cm,头围42.0 cm,均小于健康同龄儿童的2个标准差。神志清楚,精神反应好,营养不良,发育落后,不能独走,不会说话,易激惹、好动,肘部及后背皮肤多毛,特殊面容,眉毛浓密、小眼裂、长睫毛、宽眼距、塌鼻梁、球状鼻、薄上唇,骶窝(图1),手足无畸形,四肢肌力、肌张力正常,腱反射均正常,病理征均阴性。心肺腹未见异常。辅助检查:Gesell智力测验示适应性82分,大运动83分,精细动作86分,语言83分,个人-社交81分,均值83分,边缘状态;双眼屈光度检查正常;颈椎CT及全段脊髓磁共振成像(MRI)均正常;肾脏彩超示马蹄肾,右肾盂充盈;头颅MRI示胼胝体压部短,颞极蛛网膜下腔宽,侧脑室后部变尖,枕骨大孔似略窄;心脏彩超、心电图正常;血串联质谱及尿有机酸分析未见异常;甲状腺功能五项、免疫球蛋白(Ig)系列、CD系列、血氨、血乳酸、血常规、肝肾功能、电解质、凝血功能、红细胞沉降率、补体、皮质醇、促性腺皮质素未见异常。外周血染色体核型分析:46,XY。微阵列比较基因组杂交(Array CGH)检测:正常。通过全外显子组基因测序发现患儿携带KMT2A基因第27外显子c.7071delC杂合缺失移码变异,导致蛋白质改变为p.S2358Lfs*18,父母未携带,为新生变异,姐姐为野生型。查阅PubMed、人类基因突变数据库(HGMD)专业版及疾病相关基因变异(CLINVAR)数据库,该变异国内外均未见报道(图2)。在100例无亲缘关系的健康人群中亦未筛查到此变异。根据ACMG序列变异解读标准与指南对该变异进行分析为致病性变异[5]:(1)该变异为移码变异,属于功能缺失变异(PVSl);(2)新发变异,患儿父母未检出该变异(PS2);(3)该变异位于热点突变区域(PM1),在外显子测序项目(ESP)、外显子聚集联合(EAC)及千人数据库中的等位基因频率为0(PM2);(4)患儿临床症状与赖氨酸甲基转移酶2(KMT2A)基因变异所致WSS表型高度相符(PP4),Mutation Taster软件预测为有害、保守性分析变异位点高度保守(PP3),综上分析,判定KMT2A基因变异c.7071delC为致病变异。本研究通过医院医学伦理委员会批准(批准文号:2014-10),患儿监护人知情同意,并签署知情同意书。
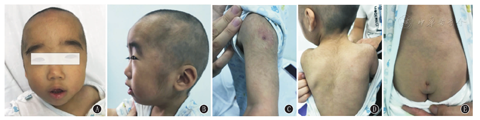
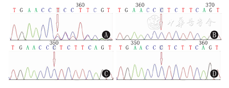
WSS是一种由KMT2A基因突变导致的常染色体显性遗传病,目前文献共报道64例[4,6,7,8,9,10,11,12,13],其中男38例,女26例,共发现KMT2A基因变异57个,无义变异23个,移码变异14个,错义变异16个,剪接位点变异3个,大片段缺失1个。3个家系先证者的变异从有症状母亲遗传获得,1个家系先证者从无症状外周血嵌合体变异的父亲遗传获得,其他均为新生变异,未发现热点变异或创始人变异,无明确基因型-表型关系。本病极为罕见,目前中国人群中仅2个家系3例患者报道[11,14],其中1例无详细临床表型描述,本例为国内详细报道的中国儿童WSS。本病主要临床表现[2,3,6]:(1)精神运动发育迟滞,其中100%的患儿均出现发育迟滞,语言发育迟滞较运动发育迟滞更明显,平均独走年龄为2岁。(2)生长迟缓,如身材矮小,身高<-2 SD,营养不良,体质量<-2 SD;部分患者有宫内生长迟缓。(3)多毛,如浓眉毛、长睫毛、肘部多毛、背部多毛、下肢多毛。(4)特殊面容,有小头畸形、塌鼻梁、宽眼距、小眼裂、薄上唇、长人中、低耳位。(5)其他神经系统表现,如肌张力减低,康复训练部分患儿肌张力可明显改善或恢复正常,半数患者肌张力减低持续存在;喂养困难:畏食、挑食;行为异常,如自我和异性攻击、注意力不足、多动症、焦虑、刻板印象、低挫折耐受性;大脑胼胝体发育不全或过细;抽搐发作。(6)不同程度骨骼异常,如骨龄落后或早熟、牙齿早熟、骶窝、足趾短或弯曲、锥形手指、颈椎畸形、肋骨畸形。(7)眼科异常,如屈光不正(远视、散光、近视),斜视,上睑下垂,少数患儿出现泪道瘘或狭窄,个别患儿出现视网膜异常、动眼神经麻痹。(8)生长激素缺乏。(9)泌尿生殖道畸形或先天心脏轻度发育异常。本病最初报道是以身材矮小、多毛和特殊面容,伴精神发育迟缓为特征性表现,随着高通量二代测序技术的发展,2012年其致病基因被发现,截至目前包括本例共65例被报道,患儿均出现精神运动发育迟滞,少数患儿并未同时出现身材矮小、多毛表现,特殊面容也缺乏特异性,因此考虑WSS的临床表现具有明显异质性。WSS需与同样可表现为特殊面容、生长迟缓及智力障碍的Kabuki综合征[13]和Cornelia de Lange综合征[15]鉴别,Kabuki综合征又称"歌舞伎脸谱综合征",亦是组蛋白修饰相关基因KMT2D和KDM6A突变致病,其特殊面容多为长眼睑、外侧眼睑外翻、弓状眉、眉外侧1/3稀疏、睫毛细长,皮纹异常较常见。Cornelia de Lange综合征的特殊外貌主要表现为连眉及弓状眉、长睫毛、长且外凸的人中、小或方形的下颚,常伴肢体缺陷,其致病基因有5个:NIPBL、SMC1A、SMC3、RAD21和HDAC8,但WSS多表现为广泛多毛、小眼睑,且身材矮小、生长迟缓在WSS中更突出和常见,临床表现不典型的患儿可通过基因检测协助鉴别明确诊断,本例全外显子基因检测未发现Kabuki综合征致病基因KMT2D和KDM6A变异,也没发现Cornelia de Lange综合征致病基因NIPBL、SMC1A、SMC3、RAD21和HDAC8存在变异。临床考虑WSS患儿建议完善以下检查:(1)脊椎MRI和CT(寻找椎动脉阻塞和椎间盘异常);(2)免疫学检查(Ig系列、CD系列等);(3)生长激素测定(如果出生后生长迟缓);(4)眼科检查(眼科查体、眼底、屈光度、视力、泪道);(5)耳鼻喉科检查;(6)心脏彩超;(7)腹部彩超;(8)脑电图和头颅MRI检查(有神经系统表现的患者,进行评估和随访);(9)基因检测[6]。本例患儿出生时身高、体质量、头围均在正常范围,无宫内发育迟缓表现,出生后即发现浓眉毛、长睫毛、肘部及后背多毛表现及小眼裂、宽眼距、骶窝的特殊外貌,随着年龄增长,特殊外貌及生长迟缓表现逐渐明显,塌鼻梁、球状鼻、薄上唇及身材矮小、营养不良、牙齿早熟,伴马蹄肾、右肾盂充盈及大脑胼胝体压部短表现,基因检测发现KMT2A基因突变明确诊断。
KMT2A基因位于11号染色体q23.3,长87 kb,共36个外显子,编码转录辅助激活子赖氨酸甲基转移酶2A,该蛋白包括3 969个氨基酸,从3′N端至5′C端共有8个保守性功能结构域,分别为结合DNA的AT-hooks结构域、富含半胱氨酸的coiled coil结构域、植物同源结构域、Zinc finger结构域、BROMO结构域、FYRN结构域、FYRC结构域和SET结构域[6]。KMT2A蛋白通过C端保守性SET结构域催化组蛋白H3赖氨酸4(H3K4)甲基化,介导染色质修饰调控表观转录激活过程,在早期发育和造血过程中对基因表达起重要调控作用。KMT2A在各个组织器官广泛表达,特别是心脏、肺、脑、T淋巴细胞和B淋巴细胞,KMT2A基因突变可导致髓系/淋巴样或混合系白血病[16]、原发性纵隔大β细胞淋巴瘤[17]及WSS,但目前尚未见报道同时出现2种表型的患者,可能与WSS患者随访年数尚短有关,建议WSS患者定期监测血常规、纵隔彩超以便早期发现异常,尽早干预。有研究报道3例WSS患者伴先天免疫缺陷,主要表现为出生后反复呼吸道或泌尿系感染,外周血IgG、IgA和IgM严重降低,CD系列正常[8,12]。本例患儿出生后外周血白细胞升高明显,CRP升高,予抗感染治疗后降至正常,10月龄时接种卡介苗,之后局部皮肤红肿及白色丘疹,持续4个月至今未消退,需注意先天免疫缺陷,但本例无反复感染病史,血Ig系列和CD系列均正常,需定期监测免疫指标。本例患儿携带的KMT2A基因第27外显子c.7071delC杂合移码变异,可通过导致蛋白质翻译截短和功能缺失而致病,为未见报道的新变异,致病机制需进一步研究。
本病目前尚无特效治疗方法,均为对症治疗,精神运动发育迟滞、肌张力减低患者需进行语言或运动康复训练。文献报道生长激素缺乏患者占50%(6/12例),但无论生长激素水平正常或降低,予身材矮小患者生长激素治疗均取得较好疗效,目前尚未发现应用生长激素治疗后合并肿瘤发生患者,需定期监测血常规、纵隔彩超并进行大样本长期随访研究评估其风险[6]。喂养困难或消瘦小婴幼儿建议高热卡奶粉喂养。脊椎CT检查出现椎骨畸形导致脊髓受压者,需进一步完善脊髓MRI,尽早手术解除脊髓压迫。眼睑下垂、小眼裂、斜视、泪道异常可行眼科手术纠正。屈光不正可完善屈光度检查佩戴眼镜纠正。心脏畸形如动脉导管未闭、卵圆孔未闭多为轻度,若未出现心功能障碍,可随访观察,多数患者随年龄增长可自愈。伴癫痫发作患者予抗癫痫药物治疗[12]。
总之,WSS是一种极为罕见的常染色体显性遗传病,对于生长迟缓、发育落后、多毛及特殊面容患者除考虑进行染色体数目或拷贝数检测外,还需考虑检测与染色体修饰重构相关的基因,全外显子组测序技术对此类疾病的诊断和新基因的发现提供了很好的检测方法,可为患儿家庭生育下一胎提供遗传咨询。
所有作者均声明不存在利益冲突

























