
建立一种低氧所致支气管肺发育不良相关肺动脉高压(BPD-PH)动物模型。
无特定病原体级C57BL/6雌雄小鼠合笼交配后,将雌鼠单独饲养,按随机数字表法分为常氧组和低氧组,常氧组母鼠所产小鼠直接置于常规室内环境饲养,低氧组母鼠所产小鼠12 h内一起放置在120 mL/L氧体积分数环境中饲养14 d。记录小鼠体质量增长、死亡率,在实验第14天获取小鼠心肺组织进行免疫荧光染色,肺组织和心脏苏木精-伊红(HE)染色,采用Western blot法对心肺组织形态、血管形态和血管内皮生长因子(VEGF)蛋白表达进行检测。
常氧组和低氧组的死亡率分别为11.8%、47.3%,造模结束体质量分别为(12.40±2.33) g、(5.50±0.32) g,差异均有统计学意义(χ2=13.38,t=20.50,均P<0.01)。与常氧组比较,低氧组右心肥厚指数升高[(96.00±0.15)%比(40.40±4.00)%,t=41.67,P<0.01];肺部形态学符合BPD的病理学改变:与常氧组比较,低氧组肺泡腔变大融合、肺泡间血管数减少,肺泡间隔断裂增厚,肺泡放射计数(RAC)减小(19.73±2.33比10.90±1.85)和平均肺泡间隔内衬(MLI)增厚(33.2±4.33比58.70±7.27),差异均有统计学意义(t=16.27、9.53,均P<0.01)。HE染色和免疫荧光染色提示低氧组肺血管增厚、平滑肌增殖、内膜粗糙;Western blot结果显示低氧组肺组织VEGF蛋白表达水平较常氧组明显降低(0.41±0.4比1.19±0.08),差异有统计学意义(t=15.10,P<0.01)。
低氧所致小鼠BPD-PH模型符合BPD-PH的肺血管重构,导致肺血管阻力升高,最终致右心室肥厚的病理生理过程。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
肺动脉高压(pulmonary hypertension,PH)是指由多种原因引起的肺动脉压力超过一定界值的一种血流动力学状态,最终导致右心负荷增大和右心功能不全。PH涵盖的疾病种类复杂多样,在疾病种类、发病机制、临床特点和治疗方案方面,儿童与成人PH均有差异。儿童PH可发生于导致肺泡及肺血管发育过程的几个关键阶段,包括胎儿期和新生儿期[1,2] 。
支气管肺发育不良(bronchopulmonary dysplasia,BPD)是早产儿接受呼吸机辅助通气和/或长时间氧疗后出现的慢性肺部疾病。研究显示,存活BPD患儿合并PH概率可高达30%,甚至更多,病死率高达48%以上[3,4,5]。BPD的病因复杂,其主要病理特征为肺泡和肺血管发育不良,表现为远端微血管丛结构形态异常和气腔表面毛细血管分布减少[6,7,8] 。在此类患者中,即便肺血管压力基线较低的患者,轻度缺氧也会引起肺动脉压力显著升高,这可以解释为血管密度减少及肺泡融合、肺泡间隔增厚造成缺氧状态,进一步加重肺血管阻力引起肺动脉平滑肌细胞增殖,二者互为因果最终导致PH的发生;反之,PH也可抑制血管生成和肺泡的发育,这也是BPD-PH不同于其他类型PH的特点[9,10] 。既往的BPD研究着重于患者和动物模型中肺泡分化异常和纤维化(肺间质损伤和修复),BPD动物模型目前非常成熟,而BPD-PH联合模型作为肺发育异常所致PH机制研究和药物干预较少有学者关注,因此,本研究在国内首次探索并评价以低氧环境饲养新生小鼠致BPD-PH模型,详细方法如下。
无特定病原体(SPF)级C57BL/6约7周雌雄小鼠,体质量20~25 g,购自广东省实验动物中心,于广州医科大学国家重点实验室SPF级动物房饲养,适应1周体质量为22~28 g。将雌雄小鼠按照12放置一笼,雌雄和笼后第2天阴道涂片观察有无交配痕迹,将有交配痕迹的雌鼠单独饲养。研究中所有动物的使用、处理操作规范等经动物使用伦理委员会审查通过。血管内皮生长因子(vascular endothelial growth factor,VEGF)蛋白抗体购自美国Abcam公司,CD31及α平滑肌肌动蛋白(α-smooth musle actin,α-SMA)抗体购自美国Cell Signaling Technology。
采用随机数字表法将12笼出生0.5 d内小鼠连同其母鼠随机分为2组:常氧组4笼,低氧组8笼,常氧组直接置于常规室内环境,低氧组放置于置于全自动缺氧箱(在每天固定早晚9点放置),以放置时间先后顺序编号1~8组,通过持续向箱内充氮气,保持氧体积分数(120±5) mL/L。箱内每天更换干燥剂,以保持干燥,第14天处死小鼠取材。出生后小鼠每3 d称重1次。
(1)标本采集:14 d后造模结束时子代小鼠称重,用30 g/L苯巴比妥钠麻醉(45 mg/kg),仰卧固定,打开腹腔,分离肝脏和膈肌,在膈肌正中位置三角区域(右心膈角)可以看到心脏搏动,沿气管分叉上方暴露主气管,剪开切口,缓慢将留置针探入,从后方连同留置针结扎支气管,以12 cmH2O(1 cmH2O=0.098 kPa)的压力沿左主支气管缓慢向左肺注入40 g/L多聚甲醛,直至全肺完全膨胀边缘变钝后将心肺组织浸泡在40 g/L多聚甲醛内。部分动物暴露心肺后直接取分离肺组织置于液氮冻存以备下一步实验。
(2)将膨胀好的肺组织置750 mL/L乙醇中,制备肺组织HE染色、免疫荧光α-SMA及CD31标记肺血管平滑肌和血管内膜。显微镜下观察肺泡放射计数(radial alveolar counts,RAC):每组各取10张切片,每张切片随机取10个视野;平均肺泡内衬间隔(mean linear intercept,MLI):随机选取2组肺组织,20倍镜下在同一水平线,每张切片随机选取8个视野,测量时避开支气管及血管,在每个视野正中心划"十"字交叉线,采用Image pro plus 6.0图像软件分析RAC和MLI值。右心肥厚指数(right-heart index,RI):心脏固定后以二尖瓣水平横向切开,石蜡包埋切片,进行HE染色,取心尖至二尖瓣三尖瓣中线部位左右心室游离壁面厚度比作为判定继发于PH的右心室肥厚,RI=右心室游离壁厚度/左心室游离壁厚度。
(3)提取液氮冻存肺组织总蛋白,测定总蛋白浓度,Western blot法检测VEGF蛋白表达水平,以GAPHD为内参蛋白。
采用SPSS 16.0软件和Prisma软件进行统计分析和作图。对于连续变量,首先进行Kolmogorov-Smirnov正态性检验,若符合正态分布,以 ±s表示。正态分布的连续变量采用双侧独立样本t检验。所有指标以双侧P<0.05为差异有统计学意义。
±s表示。正态分布的连续变量采用双侧独立样本t检验。所有指标以双侧P<0.05为差异有统计学意义。
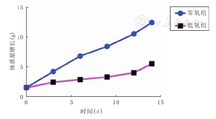
常氧组4笼母鼠共产幼鼠34只,死亡4只,死亡率为11.8%;低氧组8笼母鼠共产幼崽93只,死亡44只,死亡率47.3%,2组死亡率比较差异有统计学意义(χ2=13.38,P<0.01),提示缺氧可增加幼鼠的死亡率;待第14天实验结束时,常氧组幼鼠体质量为(12.40±2.33) g,低氧组幼鼠体质量为(5.50±0.32) g,2组比较差异有统计学意义(t=20.50,P<0.01)。造模过程常氧组大鼠体质量持续增加,与常氧组比较,低氧组体质量增加缓慢,结果见图1。
与常氧组相比,低氧组右心室明显肥厚,RI明显升高[(96.0±0.15)%比(40.40±4.00)%],差异有统计学意义(t=41.67,P<0.01),符合肺动脉高压的右心室肥厚右心室功能不全的表现。结果见图2。
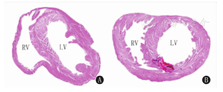
注:RV:右心室;LV:左心室 RV:right ventricle;LV:left ventricle
(1) HE染色所示:常氧组小鼠肺泡形态正常,未见肺泡融合及肺泡间隔断裂、增厚等现象,肺泡间小动脉血管壁形态、厚度均匀一致。低氧组小鼠肺泡增大,肺泡呈现融合断裂等现象,肺泡间隔增厚,肺泡间小血管数量减少,结果见图3。与常氧组比较,低氧组鼠肺组织RAC减小(10.90±1.85比19.73±2.33,t=16.27,P<0.01),MLI增厚(58.70±7.72和33.20±4.33,t=9.53,P<0.01),符合BPD的病理学改变。

注:蓝色箭头示肺泡间小血管 Interalveolar small vessels shown by blue arrow
(2)与常氧组比较,HE染色显示低氧组肺动脉管径增厚(图4),免疫荧光显示低氧组肺血管平滑肌α-SMA荧光强度增强,血管内膜CD31荧光染色显示血管内膜黏膜粗糙,提示低氧所致的BPD-PH模型中肺血管呈现平滑肌增殖现象、内膜受损现象,符合BPD-PH的形态学特征(图5)。

注:PA:肺动脉;Br:支气管 PA:pulmonary artery;Br:bronchus
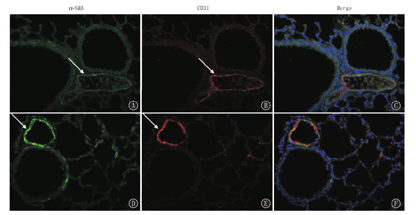
注:α-SMA:α平滑肌肌动蛋白 α-SMA:α-smooth muscle actin
与常氧组比较,Wester blot结果显示低氧组小鼠肺组织中VEGF蛋白表达水平明显降低(0.41±0.04比1.19±0.08),差异有统计学意义(t=15.10,P<0.01)。结果见图6。
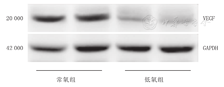
注:VEGF:血管内皮生长因子 VEGF:vascular endothelial growth factor
BPD是在1967年由挪威学者在治疗"肺透明膜病"后发现的肺部特殊病理变化并第1次描述,其病因复杂,包括慢性炎症、发育异常、氧气损伤、机械通气等。胎儿或出生后婴儿时期肺泡和远端血管的发育尚未成熟,一旦受各种致病因素的影响将导致肺血管丛损伤使其面积减少而造成结构改变,血管阻力增加。许多学者认为BPD分子生物学机制有可能与打断或者干扰正常肺部发育信号,尤其是损害肺血管丛发育和肺泡发育的因素有关,对BPD的研究在某种程度上有助于理解肺发育疾病相关PH的机制:血管生长和血管信号转导通路的损伤产生高压性血管性病变和血管重塑,这一过程发生在心肺疾病和慢性低氧血症的背景下,是导致肺发育疾病相关PH发生的病理生理基础[11,12] 。BPD-PH代表一组特殊类型PH的病理生理过程,而"缺氧"为BPD-PH的关键词。另一方面,过去20余年动物实验对BPD的病理生理机制的研究,其结果也成功转化为实际的临床应用,如预先使用糖皮质激素、肺泡表面活性物质等大大提高了BPD患儿的生存率,加之早产儿医护技术的提高,使得BPD的存活率逐年提高,而PH成了BPD患儿不可回避的问题之一[1,12]。而事实上,BPD动物模型目前已经非常成熟,包括氧损伤、机械损伤、炎症损伤等,由于BPD目前任何一种模型都不能完全模拟人类BPD疾病的特点,均存在一定缺陷,如高氧损伤所致的动物模型所需要氧体积分数一般在900 mL/L以上才能呈现相应病理损伤,而实际上新生儿BPD患者氧体积分数随着病情波动而变化,并尽量在保证基本供氧的前提下减少高体积分数氧对肺组织的持续氧化应激损伤[13,14]。基于以上因素,低氧性BPD-PH动物模型的建立有助于深入研究肺发育性疾病相关PH的发病机制。人类肺器官发育与分化大致可以分为5个阶段,包括胚胎期、假腺期、微管期、囊泡期和肺泡期,其中肺泡期可延长至3~8岁后达到肺器官完全成熟。早产儿肺发育阶段大致可跨越微管期至肺泡期,因此在此阶段各种理化因素均可影响肺部正常发育,所造成的打击引起的损伤修复病变可持续至出生后较长一段时间内(3~8岁)[13]。新生小鼠出生0~5 d仍处在囊泡期、出生5~28 d处于肺泡期,相当于人类胎儿孕后期和出生后至学龄期(孕32周至出生后8岁),在此时间段观察低氧对小鼠的囊泡期和肺泡期进行打击符合胎内缺氧和出生后3岁时BPD导致的缺氧状态,鉴于成人低氧性PH动物模型的氧体积分数为100 mL/L,本课题组在预实验探索过程中100 mL/L低氧饲养新生小鼠死亡率超过80%,后改为以120 mL/L氧气对新生小鼠进行饲养14 d,死亡率控制在50%以下,并成功诱导类似于BPD样的肺部损伤,表现为肺泡融合断裂,肺泡间隔增厚,肺泡间毛细血管数量减少,体现了低氧导致肺泡发育停滞和形态异常,肺血管丛损伤;BPD-PH组幼年小鼠体质量增长缓慢,死亡率高,也符合临床实际中BPD-PH对患儿的不良影响。研究证明,VEGF水平与BPD的发生和严重程度相关,因正常的肺发育过程中的肺泡分化依赖于血管生成[15],而VEGF表达是血管生成的重要标志[16,17],本研究选用VEGF作为BPD的观察指标之一,结果发现在低氧小鼠肺组织VEGF降低,结合HE染色的肺组织小血管数量减少,体现低氧导致肺血管生成障碍的特征。同时RI明显升高,右心室肥厚,HE染色及免疫荧光染色提示肺血管的平滑肌增厚,内膜受累,符合BPD相关PH的肺血管重构导致肺血管阻力升高最终致右心室肥厚的病理生理过程。
综上,本研究对低氧性BPD-PH动物模型制备过程和评价进行描述,通过120 mL/L氧体积分数饲养新生小鼠,制备出稳定性好的BPD-PH模型,为进一步探讨BPD-PH的病理生理机制奠定了动物模型基础。
所有作者均声明不存在利益冲突

























