
21-羟化酶缺乏症(21-OHD)是一种常染色体隐性遗传性疾病,由于肾上腺类固醇激素合成过程中21-羟化酶缺乏或活性减低导致醛固酮、皮质醇合成减少,肾上腺来源雄激素增多。近几年肾上腺雄激素合成的"后门"代谢途径及肾上腺的特异性11-氧合C19类固醇在21-OHD的作用被认识,一些特异性的肾上腺来源的雄激素正逐渐成为21-OHD诊断、治疗和监测的指标。在原有糖皮质激素及盐皮质激素替代治疗基础上,新的药物和治疗方法被逐渐研发和应用于临床。现就21-OHD诊断、治疗及监测的最新进展进行阐述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
先天性肾上腺皮质增生症(CAH)是一组由于肾上腺皮质激素合成酶基因突变导致酶活性缺乏或减低,肾上腺皮质激素合成减少,对下丘脑-垂体-肾上腺轴(HPA轴)负反馈作用减弱,促皮质素(ACTH)分泌增多,继发肾上腺皮质增生引起的一组疾病。最常见的酶缺陷是21-羟化酶缺乏,约占CAH的95%,其次为类固醇生成的急性调节蛋白、17α-羟化酶/17,20碳链裂解酶、11-β羟化酶等。总发病率为1/18 000~1/14 000[1]。21-羟化酶缺乏症(21-OHD)是由于编码21-羟化酶的基因CYP21A2突变导致,为常染色体隐性遗传,基因突变影响酶活性,依据残留酶活性的差异,临床可表现失盐型、单纯男性化型及非经典型。近几年肾上腺雄激素合成的"后门"代谢途径及肾上腺的特异性11-氧合C19类固醇在21-OHD的作用被认识,一些特异性的肾上腺来源的雄激素正逐渐成为21-OHD诊断、治疗和监测的指标。在原有糖皮质激素及盐皮质激素替代治疗基础上,新的药物和治疗方法被逐渐研发和应用于临床。现就21-OHD诊断、治疗及监测的最新进展进行阐述,以提高临床医师对21-OHD的诊治水平。
21-羟化酶由位于第6号染色体短臂6p21.3的CYP21A2基因编码(MIM# 613815;GenBank ID 1589),该基因包含10个外显子。CYP21A2位于人类白细胞抗原Ⅲ类基因区,除CYP21A2及与其高度同源但无活性的伪基因CYP21A1外,该区域同时包含编码血清补体C4的C4a、C4b基因,编码丝氨酸/苏氨酸激酶19的RP1基因,及编码肌腱蛋白X的TNXB基因等,它们彼此存在部分重叠且串联分布,形成RCCX结构(RP1-C4-CYP21A2-TNX)。无功能的CYP21A1位于CYP21A2基因上游,二者高度相似,仅有87个或88个碱基存在差异,与CYP21A2基因不同的15个突变导致CYP21A1基因失活。在有丝分裂过程中CYP21A2和伪基因之间发生基因交换重组,可能导致CYP21A2失活,这是造成CYP21A2基因失活的主要遗传学机制,除此之外常见的还有大片段缺失等。目前报道的CYP21A2突变有300多种,最常见的是IVS-13A/C→G突变[2]。有研究对84例21-OHD患者基因分析显示,中国南方地区也以c.293-13A/C>G点突变最为常见,其次为c.518T>A及c.1069C>T[3]。国内另一项研究显示,在中国北方地区最常见的点突变为c.518T>A[4]。CYP21A2基因型与表型存在一定相关性。研究表明在基因型最为严重的21-OHD患者中,二者相关联程度最强(预测阳性率为93.8%)[5]。其中启动子序列突变及外显子1(p.p30l)和外显子7(p.v281l)通常与非经典型CAH有关。外显子3(p.g110fs)、外显子4(p.i172n)及内含子2G突变通常与经典型CAH相关。患者的临床表型取决于相对较轻的突变类型。但也需注意部分研究发现基因型与表型存在不相符合的情况,分析可能与基因转录调控及蛋白翻译相关,也可能与肾上腺外其他酶如CYP2C19、CYP3A4介导的21-羟化作用相关[6]。
肾上腺皮质分为球状带、束状带、网状带,分别合成醛固酮、皮质醇及性激素。21-羟化酶在球状带介导孕酮转化为11-脱氧皮质酮进而合成醛固酮,在束状带介导17-羟孕酮(17-OHP)转化为11-脱氧皮质醇进而合成皮质醇。21-羟化酶活性减低可导致醛固酮、皮质醇合成受阻,反应底物孕酮、17-OHP堆积,旁路代谢途径增加,雄激素合成增多,临床表现为高雄激素血症。由于皮质醇生成减少,HPA轴负反馈减弱,ACTH分泌增多,导致肾上腺皮质增生。
既往对21-OHD雄激素的研究多集中在睾酮、雄烯二酮、脱氢表雄酮经典途径,近期对于肾上腺类固醇激素合成途径的研究提示,雄激素"后门"合成途径可能在CAH高雄激素血症中起更为关键的作用。雄激素"后门"途径又称为替代途径,是指17-OHP不通过睾酮直接生成双氢睾酮(dihydrotestosterone,DHT)的途径,其中关键酶是醛酮还原酶1C1/3[7]。正常情况下,经"后门"途径生成DHT在出生1年内逐渐减少。21-OHD患者由于17-OHP堆积,经"后门"途径生成DHT增多,在CAH的高雄激素血症中起重要作用[8]。
肾上腺特异性11-氧合C19类固醇在CAH高雄激素血症中也可能起重要作用。雄烯二酮和睾酮由11β羟化酶(CYP11β1)羟化作用产生11-羟雄烯二酮(11-OHAD)和11-羟基睾酮(11-OHT),然后分别在11-β-羟基类固醇脱氢酶2(HSD11β2)作用下还原为11-酮雄烯二酮(11-KAD)和11-酮睾酮(11-KT)。该途径既往被认为是肾上腺类固醇激素灭活途径,未予特殊重视,但近期研究表明,该途径在肾上腺合成雄激素中发挥重要作用。11-KT经5α还原酶作用生成11-酮双氢睾酮(11-KDHT)。11-KAD与雄激素受体结合能力弱,11-OHAD可部分激活雄激素受体,11-OHT及11-KT可与雄激素受体完全结合,其中11-KT活性与睾酮相当[9,10]。研究表明,21-OHD患者11-OHT、11-OHAD、11-KAD及11-KT等均较健康人群显著升高,推测其可能在21-OHD高雄激素血症中发挥重要作用[9,10]。
在肾上腺类固醇激素中被重新发掘的还有21-脱氧皮质醇(21-deoxycortisol,21-deoxy)。17-OHP在CYP11β1作用下生成21-deoxy,既往由于检测技术受限,对于21-deoxy研究较少。近期发现,在21-OHD患者中21-deoxy明显升高,与17-OHP具有高度一致性,与17-OHP不同的是在新生儿中不受胎龄等限制,因此可用于新生儿21-OHD的筛查[11],见图1。
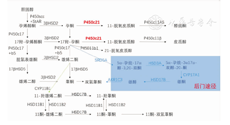
注:P450scc:细胞色素P450胆固醇侧链裂解酶;StAR:固醇激素合成急性调节蛋白;3βHSD2:3β羟基类固醇脱氢酶2;P450c17:17α-羟化酶/17,20-裂解酶;P450c21:21-羟化酶;P450c11AS:醛固酮合成酶;P450c11β/P45011b1:11β-羟化酶;b5:细胞色素b5;SRD5A:类固醇5α-还原酶;17βHSD5:17β-羟类固醇脱氢酶5;HSD3A:3α-羟类固醇脱氢酶;AKR1C3:醛酮还原酶1C3;HSD17B:17β-羟类固醇脱氢酶;HSD11B1:11β-羟类固醇脱氢酶1;HSD11B2:11β-羟类固醇脱氢酶2 P450scc:cholesterol-side-chain cleavage enzyme;StAR:steroidogenic acute regulatoryprotein;3βHSD2:3β-hydroxysteroid dehydrogenase 2;P450c17:17α-hydroxylase/17,20 lyase;P450c21:21-hydroxylase;P450c11AS:aldosterone synthase;P450c11β:11β-hydroxylase;b5:cytochrome b5;SRD5A:steroid 5-alpha reductase;17βHSD5:17β-hydroxysteroid dehydrogenase 5;HSD3A:3α-hydroxysteroid dehydrogenase;AKR1C3:aldo-keto reductase family 1 member C3;HSD17B:17β-hydroxysteroid dehydrogenase;HSD11B1:11β-hydroxysteroid dehydrogenase1;HSD11B2:11β-hydroxysteroid dehydrogenase 2
依据21-羟化酶残留活性差异,21-OHD临床分为3种类型:失盐型、单纯男性化型及非经典型。
失盐型是最严重的21-OHD表型,无残留酶活性,醛固酮、皮质醇完全缺乏,雄激素生成增多。出生后不久即可出现呕吐、拒食、脱水、代谢性酸中毒、低钠血症和高钾血症等肾上腺危象表现,女性可有外生殖器男性化,若不及时诊治,病死率高。
残留酶活性1%~2%,临床无失盐型表现,女性患者出生时即有外生殖器男性化表现,男性患者可出现假性性早熟。
也称迟发型,21-羟化酶残留酶活性20%~50%。临床表现不特异,可在儿童期或青春期甚至成人期发病,出现阴毛早现、性早熟、月经紊乱、不孕等。
出生后发现外生殖器发育异常、吐奶、拒食、体质量不增的新生儿及性早熟、原发性闭经、月经紊乱、不孕且伴外生殖器色素沉着的儿童或成人均需注意21-OHD的可能。
失盐型21-OHD患者早期可出现肾上腺危象,新生儿筛查可降低病死率,目前已被逐渐推广。应用放射免疫法检测血清17-OHP水平是21-OHD的一级筛查方法。健康儿童出生后17-OHP可轻度升高,出生1 d迅速下降,因此建议在出生48 h后进行。早产儿17-OHP较足月儿童升高,因此需要建立不同胎龄17-OHP筛查切值。除此之外疾病状态、应激等也会导致17-OHP升高。限于检测方法,其他肾上腺类固醇激素交叉免疫反应也可导致筛查结果呈现假阳性,因此对于上述儿童需要进行二级筛查。目前推荐的二级筛查指标是采用液相色谱-串联质谱法检测17-OHP水平,早产儿建议在出生后2周及4周进行17-OHP复测。近期有研究显示,其他生物标志物如21-deoxy、尿孕三酮、尿6α-羟基四氢可的松等也可用作筛查指标[12]。其中21-deoxy在不同胎龄新生儿中无明显差异,避免了胎龄对筛查造成的影响[13]。但21-deoxy应用放射免疫法检测受交叉反应影响较大,建议采用液相色谱-串联质谱法进行检测,但由于该技术难度较高,目前尚未在临床广泛推广。其他方法如基因检测目前不推荐作为二级筛查指标,仅在类固醇检测不能明确诊断时建议采用。
筛查阳性患儿,或在儿童期有症状怀疑21-OHD患者,需行ACTH 1-24激发试验。于清晨8∶00-9∶00进行,静脉推注0.25 mg ACTH,极低出生体质量儿可减少为0.125 mg,检测基线值及60 min时血清17-OHP水平,如有条件,建议同时进行脱氢表雄酮、雄烯二酮、11-脱氧皮质醇、11-脱氧皮质酮水平测定,相关激素比值可用于鉴别CAH类型[14]。21-OHD诊断流程见图2。
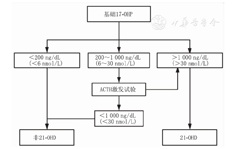
注:17-OHP:17-羟孕酮;ACTH:促皮质素;21-OHD:21-羟化酶缺乏症 17-OHP:17-hydroxypregnenolone;ACTH:adrenocorticotropic hormone;21-OHD:21-hydroxylase deficiency
21-OHD治疗的目的是纠正糖皮质激素、盐皮质激素缺乏及抑制ACTH和肾上腺来源的高雄激素过多分泌。治疗包括药物及手术治疗。
21-OHD患者糖皮质激素替代治疗的目的是纠正皮质醇缺乏,但为抑制ACTH及雄激素过多分泌,替代剂量通常高于生理需要量。处于生长发育期的21-OHD患者,为减少对生长的影响,建议应用氢化可的松进行糖皮质激素替代治疗,推荐剂量为每日10~15 mg/m2,分3次服用。关于糖皮质激素每日服用次数及是否存在不同时间的剂量差异目前尚缺乏足够研究数据,不能得出统一结论。由于个体对药物代谢存在差异,可通过监测24 h氢化可的松药物浓度进行药物代谢动力学研究,测定个体药物达峰时间及峰浓度,进而指导药物应用[15]。对于已终止生长的儿童,可采用长效糖皮质激素进行替代治疗以改善患者依从性。
关于不同糖皮质激素替代治疗对CAH治疗效果及相关不良反应的Meta分析发现,地塞米松与氢化可的松及甲泼尼龙相比,地塞米松可最大程度抑制ACTH,在雄激素方面3种激素治疗中17-OHP和雄烯二酮水平差异无统计学意义。但同时发现,地塞米松治疗的患者骨密度较其他糖皮质激素治疗患者明显减低,体质量指数升高,提示应用地塞米松进行CAH糖皮质激素替代治疗可能增加骨质疏松发生风险,肥胖、代谢性疾病及心血管疾病等远期并发症发生风险也可能升高[16]。
尽管仅有失盐型患者具有相关失盐表现,但实际上所有的21-OHD患者均存在盐皮质激素缺乏。对于21-OHD患者,建议在儿童期加用盐皮质激素进行替代治疗,目前应用的药物为人工合成9α-氟氢可的松,剂量为0.05~0.20 mg/d,分1~2次口服。仅在低钠血症不能纠正时可短期增加药物剂量,但此时需注意9α-氟氢可的松也具有一定糖皮质激素作用,长期大量应用可能导致库欣综合征等并发症。既往研究认为,单纯男性化型及非经典型21-OHD患者能产生足够醛固酮,因此不需额外补充盐皮质激素,但这部分患者醛固酮与肾素的比值较健康人减低,服用盐皮质激素可使其恢复正常,因此建议这部分患者同时进行盐皮质激素替代治疗[17]。胎儿期肾脏盐皮质激素受体mRNA含量低,对激素反应差,随着年龄增长受体活性增强,需求逐渐减少。因此在青春期过渡至成人期,需重新评估盐皮质激素代谢情况,判断是否继续应用盐皮质激素。
对于外生殖器男性化的21-OHD患者,手术目的是去除多余的勃起组织,保存性敏感的阴蒂腺体,提供正常的尿道阴道开口,减少由于尿液在阴道或泌尿生殖道内聚集而引起的感染。对于手术时机及方式选择目前缺少长期随访研究。早期手术有助于恢复正常解剖结构、减少泌尿系统感染发生、降低患者及其父母的心理压力及缓解焦虑情绪,减少心理损伤,但研究发现早期进行手术术后发生阴道狭窄风险高。2018年美国内分泌学会21-OHD指南建议对于尿生殖窦共同开口较低的21-OHD患者应早期进行阴道成形术,尿生殖窦共同开口较高患者目前最佳手术时期选择不能确定。重度男性化患者手术应慎重选择,以避免性别认同障碍发生。
青春期21-OHD女性患者由于雄激素水平过高,可考虑同时加用雄激素受体拮抗剂以减轻高雄激素血症,但失盐型患者加用螺内酯需慎重。屈螺酮(优思明-屈螺酮炔雌醇片)可抑制肾上腺及卵巢来源雄激素,且不影响皮质醇合成、血清电解质浓度、血压及肾素活性,也可考虑应用,但目前缺乏相关研究。
21-OHD患者若合并中枢性性早熟可考虑应用促性腺激素释放激素类似物治疗,以抑制中枢性腺轴改善终身高。但治疗开始时机较为重要,若骨龄延迟明显,该治疗对于抑制生长速率无显著改善[18]。
联合应用芳香化酶抑制剂也能够减缓骨龄进展,但需注意芳香化酶抑制剂治疗可导致血清17-OHP、雄烯二酮水平升高,这给药物治疗监测带来一定困难,需要结合临床症状经验性调整药物剂量[19]。
糖皮质激素泵:可模拟皮质醇生理分泌,减少相关治疗不良反应。目前2期临床试验已完成,参与试验的8例CAH患者在试验的6个月中监测17-OHP、雄烯二酮和孕酮明显减低,生活质量评分较前显著提高,且减少了氢化可的松用量[20]。
糖皮质激素缓释剂:氢化可的松缓释剂通过多环节控释,已达到药物多环节缓释目的,可减少药物服用次数,在原发性肾上腺皮质功能不全患者中研究显示可显著改善体质量指数、降低糖化血红蛋白、提高生活质量[21]。在CAH患者中,相对于常规糖皮质激素治疗,其能够降低17-OHP及尿雄激素代谢产物,提示在改善雄激素过多中可能具有优势[22]。
NBI-77860:选择性促肾上腺皮质激素释放因子1型(CRF-1)拮抗剂。1期临床试验8例受试者中有6例出现ACTH、17-OHP剂量依赖性降低。与安慰剂相比,300 mg NBI-77860应用使ACTH和17-OHP平均降低43.0%和0.7%,600 mg NBI-77860应用使ACTH和17-OHP平均降低41.0%和27.0%[23]。
ATR-101:即酰基辅酶A胆固醇酰基转移酶1(ACAT-1)选择性抑制剂,是一种新的小分子治疗药物,ACAT1催化来自胆固醇和长链脂肪酰辅酶A的胆固醇酯的形成,ATR-101可选择性抑制肾上腺ACAT1活性,减少肾上腺皮质类固醇激素合成,可用于治疗肾上腺皮质癌、库欣综合征[24,25],目前该药用于治疗21-OHD的2期临床试验已完成(NCT02804178),治疗目标主要用来降低肾上腺来源的雄性激素(17-OHP、雄烯二酮、脱氢表雄酮等)水平和防止肾上腺增生,但相关结果尚未见发表。
醋酸阿比特龙:是17α羟化酶/17,20碳链裂解酶的不可逆抑制剂,是睾酮合成所需的关键酶,可有效抑制21-OHD患者的高雄激素血症。1期临床试验已证实,醋酸阿比特龙能够显著降低21-OHD患者血清雄烯二酮水平,且呈剂量依赖性[26]。但该药导致上游类固醇累积的长期后果尚不清楚,与酶结合不可逆,如不能及时补充糖皮质激素,过多的ACTH可促进新的酶合成释放,造成药物逃逸。目前该药的2期临床试验已申请,但尚未招募(NCT03548246)。
基因治疗:2018年发表的一项研究发现,以腺病毒相关病毒(AAV)为载体基因治疗21-OHD小鼠,单次静脉注射AAV血清型rh.10转基因质粒(AAVrh.10-21OH-HA)可以有效地转导肾上腺皮质细胞,使21OH-HA表达和分泌恢复,恢复正常的类固醇生成,但治疗有效期仅8周。因此,单一通过腺病毒载体进行的、以补偿功能性基因拷贝为目的的基因疗法只能暂时性缓解21-OHD,因为位于肾上腺的干细胞对肾上腺皮质细胞具有不断更新替代作用,所以通过基因改变肾上腺皮质干细胞群的策略可能是新的研究方向[27]。
在21-OHD治疗过程中,过量的糖皮质激素治疗可导致婴儿期不可恢复的生长受限及医源性库欣综合征等,激素剂量减少则会增加肾上腺危象发生风险,并且不能有效抑制肾上腺来源的雄激素。因此临床应用时必须密切监测相关指标,在充分抑制肾上腺来源雄激素水平、控制男性化症状的前提下尽量减少激素用量。需要定期关注相关临床症状,如阴毛生长、阴茎生长、生长速率异常改变、大汗腺异常气味及嗜盐和肾上腺危象表现,监测身高、体质量、血压。类固醇激素监测在21-OHD治疗中起重要作用。17-OHP水平能有效地反映ACTH抑制水平,是传统监测指标。17-OHP应控制在正常上限,但考虑到样本取样时间不同、实验室参考范围不同及个体差异,无法规定治疗监测过程中17-OHP具体界值。
既往认为雄烯二酮、脱氢表雄酮、硫酸脱氢表雄酮及睾酮能够反映21-OHD雄激素代谢,但研究显示上述类固醇激素并不能完全反映疾病的严重程度及评估治疗反应[28]。肾上腺特异性11-氧合C19类固醇及雄激素"后门"途径相关激素逐渐被纳入视野。但是利用传统的放射免疫法检测上述类固醇存在困难,目前仅有少量研究报道,不能得出有效结论,需进一步探索。
2019年发表的一项研究显示,利用液相色谱法检测尿液皮质醇代谢产物可用于监测CAH激素治疗的疗效,研究发现尿液中所有雄激素代谢物的Z值应控制在0.512(准确率为66.2%,敏感性为57.1%,特异性为74.4%,阳性预测值为66.7%,阴性预测值为65.9%),升高提示雄激素过量。尿液中四氢皮质醇排泄量Z值为0.163表示过度治疗(准确率为79.6%,敏感性为40.0%,特异性为94.9%,阳性预测值为75.0%,阴性预测值为80.4%)[29]。
视网膜作为大脑的一部分,男性视网膜厚度明显高于女性。研究发现,21-OHD女性患者视网膜厚度明显高于健康女童,提示视网膜厚度可作为21-OHD女性过度暴露于雄激素的形态学标志,为CAH治疗监测提供了新思路[30]。
>2岁儿童还应进行骨龄监测,骨龄进展至青春前期建议进行促性腺激素释放激素激发试验评估性腺发育程度。男性21-OHD应定期监测睾丸超声,以及时发现肾上腺残余瘤。
所有作者均声明不存在利益冲突

























