
总结1例PLPBP基因突变导致吡哆醇依赖性癫痫(PDE)患儿的临床特征及基因突变特点,并进行文献复习。
对2017年12月就诊于北京大学第一医院儿科的1例PDE患儿的临床及基因检测进行分析。以"吡哆醇依赖性癫痫" 、"pyridoxine-dependent epilepsy" 、"PLPBP" 、"PROSC"为关键词查阅在线人类孟德尔遗传数据库、PubMed数据库、中国知网及万方数据库从建库至2019年1月相关文献,并对国内外报道的19例PLPBP基因突变所致PDE确诊病例进行汇总分析。
女,1岁3个月,出生7 d反复癫痫发作,抗癫痫药物未能控制,给予吡哆醇后发作控制。9个月时,仅吡哆醇单药治疗下日常发作控制良好,遇有发热性疾病时会复发,患儿自发病以来发育基本正常。全外显子组测序显示PLPBP基因有2个复合杂合突变,c.119C>T(p.P40L)和c.207+1G>T。对国外3篇文献报道的19例PLPBP基因突变导致PDE确诊病例和本例汇总分析显示,20例均于新生儿期出现难治性癫痫,26.3%(5/19例)为早产儿,30.0%(6/20例)孕期胎动异常,63.2%(12/19例)有不同程度的发育落后,1例于4.5个月时死亡。癫痫发作形式多样,包括全面强直阵挛发作,肌阵挛发作,强直发作,痉挛发作,局灶性发作。58.3%(7/12例)出生时乳酸升高,47.4%(9/19例)脑电图表现为爆发抑制,31.6%(6/19例)为多灶性癫痫样放电,5.3%(1/19例)为正常。仅30.0%(6/20例)的患儿经吡哆醇单药控制发作。
PLPBP基因突变所致的PDE患儿常为新生儿期出现癫痫发作、早产儿发生率较高、孕期可出现异常胎动。多数出生后有乳酸升高,脑电图爆发抑制较为常见。仅少数可被吡哆醇单药控制,多数患儿发育落后。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
吡哆醇依赖性癫痫(pyridoxine-dependent epilepsy,PDE)是一种常染色体隐性遗传性疾病,通常在新生儿期或婴儿期出现难治性癫痫发作,其特征是癫痫发作不能被抗癫痫药物控制,但可被大剂量吡哆醇控制或明显改善,一旦停用吡哆醇,癫痫发作会在1~51 d复发[1]。该病最早由Hunt等[2]于1954年首次报道,至2019年1月,国内外报道200余例[3]。2006年PDE的致病基因乙醛脱氢酶7家庭成员A1基因(ALDH7A1)被确定[4]。2016年,一种新的致病基因PLPBP(pyridoxal-5′-phosphate-binding protein,之前又称PROSC)被确定,PLPBP编码的磷酸吡哆醛(PLP)结合蛋白参与细胞内游离PLP稳态调节[5]。目前国外共3篇文献报道了19例PLPBP基因致PDE病例[5,6,7],国内尚未见相关的病例报道。现对北京大学第一医院儿科确诊的1例PLPBP基因突变致PDE患儿的临床及PLPBP基因突变特点进行总结,并进行文献复习及分析,以提高对本病临床及遗传学特点的认识。
患儿,女,3个月,因"频繁抽搐"于2017年12月就诊于北京大学第一医院儿科神经门诊。体格检查:患儿病程中多次测量头围均正常,四肢肌张力稍高,双侧膝反射可正常引出,巴氏征阴性。家族史:家族中无类似病史,有一个姐姐,体健。患儿系第3胎、第2产,足月顺产,母孕期患有妊娠期糖尿病,患儿出生时低血糖,予对症处理后血糖恢复正常。患儿出生第7天无明显诱因出现惊厥,表现为双手握拳、四肢抽动,每次持续30 s~1 min,10 min内丛集性发作7~8次。抽搐当天于外院住院治疗,予苯巴比妥(PB)治疗无效,添加左乙拉西坦(LEV)口服,并给予吡哆醇静脉滴注(100 mg/d)联合治疗,24 h内发作控制,共静脉滴注吡哆醇3 d,发作控制6 d后出院,院外继续口服LEV,但未服用吡哆醇。出院后3 d再次出现癫痫发作,表现为双眼球快速转动、双眼凝视、表情惊恐、四肢抽动或双上肢上抬,伴有哭闹,持续20~40 min,每日发作2~3次,于门诊调整LEV剂量,症状改善不明显。患儿3个月时来北京大学第一医院住院治疗,入院后第1天给予托吡酯(TPM)口服后发作未改善,第2天给予吡哆醇100 mg/d静脉滴注,用药2 d后发作控制,共静脉滴注5 d,癫痫发作控制4 d后出院。出院后常规口服吡哆醇60 mg/d,并同时服用TPM和LEV。患儿7个月时,在癫痫发作控制的情况下逐渐减停抗癫痫药物,经2个月减完,近6个月仅口服吡哆醇90 mg/d,日常发作控制良好,期间遇有2次发热性疾病时,为防止癫痫发作复发,家长在发热第1天即将吡哆醇剂量加倍至180 mg/d口服,但热程中仍各出现了1次癫痫发作,分别持续20 min及5 min,均同时给予地西泮静脉推注后发作终止。本研究通过北京大学第一医院医学伦理委员会批准(批准文号:2016-1135),患儿监护人均知情同意,并签署知情同意书。
行血液生化、血氨基酸、血酯酰肉碱谱、尿有机酸、脑电图(EEG)及头颅磁共振成像(MRI)检查,并进行临床粗略智力评估。
癫痫相关基因包检测:委托康旭医学检验所进行,采用目的基因靶向捕获二代测序技术,基因包内包含470个已知的癫痫相关致病基因(包括ALDH7A1及PNPO基因,但不包括PLPBP基因)。家系全外显子组基因测序(含4 000种单基因遗传病基因):委托智因东方医学检验所进行,并对发现的可能致病突变,进一步采用Sanger测序法进行验证,以明确突变来源。
辅助检查:血常规及生化正常,血、尿代谢筛查正常,乳酸3.6 mmol/L,头颅MRI正常。EEG检查:对本例患儿出生17 d~10个月中6次系列EEG分析显示,早期用吡哆醇治疗时EEG仅少量放电;在仅用抗癫痫药治疗时,EEG表现为多灶性及广泛性放电,并监测到局灶性发作,甚至痉挛发作混合局灶性发作接近持续状态;在用吡哆醇静脉滴注或口服治疗后,EEG没有立即恢复正常;吡哆醇撤药后,癫痫发作复发且伴随EEG加重;但在吡哆醇单药长期维持治疗后癫痫发作控制及EEG恢复正常并维持(表1)。患儿起病年龄小,起病前发育未发现明显异常。随访至1岁3个月,癫痫发作已持续控制6个月,会叫"爸爸、妈妈",能独走。
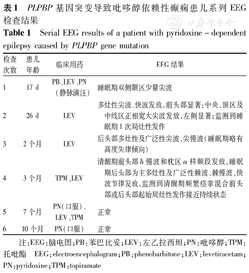
PLPBP基因突变导致吡哆醇依赖性癫痫患儿系列EEG检查结果
Serial EEG results of a patient with pyridoxine-dependent epilepsy caused byPLPBP gene mutation
PLPBP基因突变导致吡哆醇依赖性癫痫患儿系列EEG检查结果
Serial EEG results of a patient with pyridoxine-dependent epilepsy caused byPLPBP gene mutation
| 检查次数 | 患儿年龄 | 临床用药 | EEG结果 |
|---|---|---|---|
| 1 | 17 d | PB、LEV、PN(静脉滴注) | 睡眠期双侧颞区少量尖波 |
| 2 | 26 d | LEV | 多灶性尖波、快波发放,前头部显著:中央、顶区及中线区正相宽大尖波发放,左侧显著;监测到睡眠期1次局灶性发作 |
| 3 | 2个月 | LEV | 后头部多灶性及广泛性尖波、尖慢波(睡眠期略有高度失律倾向) |
| 4 | 3个月 | TPM、LEV | 清醒期前头部δ慢波和枕区α样频段发放,睡眠期后头部为主多灶性及广泛性棘波、棘慢波、快波节律发放,监测到清醒期频繁痉挛混合前头部或后头部起始局灶性发作接近持续状态 |
| 5 | 7个月 | PN(口服)、LEV、TPM | 正常 |
| 6 | 10个月 | PN(口服) | 正常 |
注:EEG:脑电图;PB:苯巴比妥;LEV:左乙拉西坦;PN:吡哆醇;TPM:托吡酯 EEG:electroencephalogram;PB:phenobarbitone;LEV:levetiracetam;PN:pyridoxine;TPM:topiramate
癫痫相关基因包检测未发现相关性异常。进一步全外显子组测序发现患儿携带PLPBP基因复合杂合突变:第2外显子c.119C>T(p.P40L)杂合错义突变(图1A)和第2内含子c.207+1G>T剪切位点突变(图1B)。父母来源验证显示,其父携带c.119C>T突变,其母携带c.207+1G>T突变。结合患儿临床表现,考虑该PLPBP基因突变具有致病性,明确诊断为PLPBP基因突变导致的PDE。
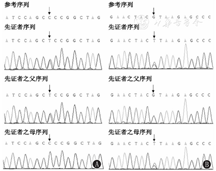
注:患儿携带PLPBP基因复合杂合突变:第2外显子c.119C>T(p.P40L)杂合错义突变(A)和第2内含子c.207+1G>T剪切位点突变(B),其父携带c.119C>T突变,其母携带c.207+1G>T突变;箭头示突变位点 The patient was revealed to have complex heterozygous mutations inPLPBP gene:a heterozygous missense mutation c.119C>T (p.P40L) in exon 2 was inherited from the father (A) and a splicing site mutation c.207+1G>T in intron 2 was inherited from the mother (B);The arrow shows the mutation site
国外3篇文献报道19例PLPBP基因突变导致PDE确诊病例,结合本例共20例。其中男10例,女10例。
26.3%(5/19例)的患儿为早产儿,病例均为出生24 h内或1个月内起病,50.0%(10/20例)出生24 h内起病,45.0%(9/20例)出生9 d内起病,1例出生1个月起病。30.0%(6/20例)孕期胎动异常,30.0%(6/20例)孕期宫内窘迫,25.0%(4/16例)出生后出现呼吸衰竭。癫痫发作表现为多种类型:全面强直-阵挛发作(45.0%,9/20例)、肌阵挛发作(35.0%,7/20例)、强直发作(35.0%,7/20例)、痉挛发作(30.0%,6/20例)、局灶性发作(5.0%,1/20例),其中1例患儿为光敏感性癫痫发作,73.7%(14/19例)发热时会诱发发作。
12例出生时检测了血乳酸,7例升高,5例正常。22.2%(2/9例)的患儿出生时有贫血,余未检测或未报道。在完成EEG检查的19例患儿中,9例为爆发抑制,1例为爆发抑制倾向,1例为不连续图形(胎龄为38+2周),6例提示多灶性癫痫样放电(1例伴背景活动异常),1例背景活动异常及局灶性放电,1例正常。完成头颅MRI检查的19例患儿中,12例正常,7例存在异常:脑回宽及脑沟浅的全脑发育不全、脑白质发育不良、左侧或双侧近额角处囊肿、胼胝体薄、脑白质深部水肿并有出血点、脑室旁(假)囊肿、内囊后肢未髓鞘化、额叶脑回发育不良、侧脑室和第三脑室轻度扩张。
20例均进行了PLPBP基因突变检测,共发现15种不同的突变位点,其中c.233C>G(3例)、c.199G>A(3例)和c.122G>A(3例)较为常见,且都为纯合突变;患儿均符合常染色体隐性遗传,其中15例为纯合突变,5例为复合杂合突变。
20例患儿均接受吡哆醇治疗,1例无效,19例经吡哆醇治疗后显示不同的疗效:完全控制发作(31.6%,6/19例),发作次数减少(68.4%,13/19例)。100%(8/8例)的患儿在吡哆醇撤药后出现复发;5例由吡哆醇转换为PLP治疗,癫痫发作均有进一步改善(文献未提及是否完全控制),2例在PLP撤药后出现反复。20例患儿中1例于4.5个月时死亡,12例有不同程度的精神行为发育迟滞,表现为言语迟缓、轻-重度学习困难、独走延迟等,7例发育正常。
PDE是一种罕见的常染色体隐性遗传性疾病,有关患病率的报道较少,且差异很大,介于1∶700 000~1∶20 000[8]。国内于2013年报道了首例ALDH7A1基因确诊病例,并随后陆续报道了一系列病例[8,9,10,11,12,13]。但因PLPBP基因发现较晚,至今国内尚未见相关病例报道。本例为国内首例通过PLPBP基因突变确诊的PDE病例。
对ALDH7A1基因突变所致的PDE患儿研究显示,男女患病比例无差异,17.9%(5/28例)有早产史,33.3%(8/24例)孕期胎动异常[1]。典型PDE临床表现为在新生儿期或婴儿期出现难治性癫痫发作。癫痫发作形式多样,可出现痉挛发作、强直发作、肌阵挛发作、全面强直-阵挛发作等[1,3,14],且不能被抗癫痫药物控制。在本研究总结的20例患儿中亦未见性别差异,26.3%(5/19例)为早产儿,30.0%(6/20例)孕期胎动异常[5,6,7],提示存在胎儿期癫痫发作可能,但因部分患儿在文献中缺少对孕期的详细描述,以上比例或许存在偏差。本研究总结的患儿中半数在出生24 h内出现癫痫发作,最晚于出生1个月起病,提示PLPBP基因突变所致PDE患儿更易在新生儿期起病。与ALDH7A1基因突变所致PDE患儿相似,患儿均在病程中出现多种发作类型,以全面强直-阵挛发作、肌阵挛发作、强直发作较为常见,且对多种抗癫痫药物(PB、LEV、VPA、TPM等)亦无反应[5,6,7]。
ALDH7A1基因突变引起的PDE中,新生儿期实验室检查可存在乳酸酸中毒、贫血、低血糖、电解质紊乱等[1,15],但无特异性提示作用;本研究总结的PLPBP基因突变所致的患儿中,高达58.3%(7/12例)出生后乳酸水平明显升高[5,6,7],从而有助于在疾病早期提示临床医师对患儿进行吡哆醇试验性治疗。本例患儿出生时患有低血糖,但因患儿的母亲患有妊娠期糖尿病,因此不能确定其低血糖是由本病还是其母亲所致。有研究报道,在ALDH7A1基因突变导致的PDE中,31.6%(6/19例)头颅MRI正常,也可为非特异性异常[1,16]。本研究总结的患儿中63.2%(12/19例)头颅MRI正常,余为非特异性异常。PLPBP基因突变患儿头颅MRI正常的比例明显高于前者,是源于2个基因本身的不同所致,还是与PLPBP基因发现较晚、病例随访时间较短有关,尚待进一步的随访和分析。本例患儿头颅MRI正常,但因病程短,远期MRI是否有变化尚需观察。
文献总结ALDH7A1基因突变所致PDE患儿的EEG显示,15.8%(3/19例)为爆发抑制图形,57.9%(11/19例)显示背景活动减慢伴多灶性或广泛性癫痫样放电,发作期EEG因发作类型不同而异,10.5%(2/19例)EEG为正常[1]。在本研究总结的PLPBP基因突变导致PDE患儿的EEG显示,47.4%(9/19例)表现为爆发抑制,31.6%(6/19例)为多灶性癫痫样放电,5.3%(1/19例)正常[5,6,7],对比可见,爆发抑制EEG在PLPBP基因突变导致PDE患儿中更为常见。一直以来,观察EEG监测下静脉注射吡哆醇的反应被认为有助于诊断或排除本病,但后期研究显示,吡哆醇静脉注射后EEG呈现非特异性反应[17]。本例患儿病程中6次EEG结果分析显示,EEG随吡哆醇治疗及撤药呈现对应性好转与恶化,当长期稳定治疗后EEG持续正常,但并非治疗后即刻显著反应。
至2019年1月,国际上共报道了15种不同的PLPBP基因突变位点,其中以c.233C>G、c.199G>A和c.122G>A较为常见[5,6,7]。本例患儿为复合杂合突变,c.119C>T(p.P40L)的致病性国际上已报道[6],c.207+1G>T剪切位点未报道,但c.207+1G>A的致病性国际上已报道,支持该突变的可能致病性。本例患儿曾进行癫痫基因包检测,但因仅包含ALDH7A1及PNPO等基因,而PLPBP基因由于发现的较晚未被涵盖,所以未能通过基因包检测确诊,由此提示临床在疾病指向性不甚明确时,最好选择家系全外显子组检测而不是癫痫基因包。Johnstone等[7]报道的12例PLPBP基因突变患儿中,4例临床分别诊断为线粒体脑病(2例)、亚叶酸反应性癫痫(1例)及伴有抗体依赖的细胞介导的细胞毒性作用缺乏的运动障碍(1例)。该4例患儿在口服吡哆醇治疗后不能完全控制发作或撤药后不会引起癫痫复发,因此,不能诊断为PDE,提示PLPBP基因突变也可能为其他类型神经系统疾病的致病基因。
有研究报道,在ALDH7A1基因突变的患儿中,72.7%(8/11例)可被吡哆醇单药控制,但患急性发热性疾病期间易复发,此时可通过增加吡哆醇剂量来预防或控制爆发性惊厥发作[18]。以上在本研究总结的患儿中均有体现,吡哆醇撤药的8例(100%)及PLP撤药的2例(100%)患儿中均有复发,但与ALDH7A1基因突变不同的是,在PLPBP基因突变导致PDE患儿中,仅30.0%(6/20例)的癫痫发作可通过吡哆醇单药控制,70.0%(14/20例)在单用吡哆醇的情况下仍有发作,需要联合抗癫痫药物治疗。在换用PLP治疗的患儿中,均表现出癫痫发作控制情况改善[5,6,7],这表明PLPBP基因突变可能不仅仅影响中枢神经系统中PLP水平,还影响了其他物质的代谢,从而引起更严重的临床表型。本研究中,73.7%(14/19例)在发热时出现癫痫复发[5,6,7],本例患儿为防止癫痫发作复发,家长在发热第1天即将吡哆醇剂量加倍口服,但仍未能阻止癫痫发作的复发,发作时给予地西泮静脉推注后终止,提示临床医师在PDE患儿患急性发热性疾病期间若增加吡哆醇剂量未能控制复发,则仍需给予镇静剂来控制癫痫发作。
既往报道中,71.4%(10/14例)的ALDH7A1基因突变所致PDE患儿表现出显著的智力残疾和发育迟缓[19],在本研究总结的病例中,63.2%(12/19例)有不同程度的智力运动发育落后[5,6,7]。本例患儿随访近6个月癫痫控制较好,智力运动发育基本正常,尚需继续长期随访观察。由本研究总结的病例可看出,c.199G>A纯合突变的患儿存在言语迟缓和发育落后,c.122G>A纯合突变的患儿发育均正常,c.233C>G纯合突变的患儿中1例死亡、2例发育明显落后[5,6,7],以上提示同一基因的不同突变位点所致预后有所不同。
所有作者均声明不存在利益冲突

























