
报道2例Ullrich型先天性肌营养不良(UCMD)患儿临床特点,初步探讨康复管理方案。2例患儿临床特点为早发型全身肌无力,以近端为著,远端关节松弛,近端关节挛缩,亦或有瘢痕形成异常及皮肤过度角化,儿童期即有呼吸功能减退,心脏不受累。下肢肌肉磁共振成像特点为大腿肌肉弥散脂肪化,单块肌肉受累边缘重、中央轻,以股外侧肌明显,呈"三明治"征,股直肌中央亦有受累,缝匠肌、长收肌、股薄肌相对受累较轻。患儿1的COL6A3基因存在c.6210+1G>C杂合突变,该变异为新发变异,患儿2为COL6A1基因Exon9 c.850G>A (p.Gly284Arg)杂合突变,为错义突变。目前尚无针对该病的特异性药物,可遵循缓慢进展的神经肌肉疾病康复管理原则行综合康复管理。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Ullrich先天性肌营养不良(UCMD)是一种常染色体隐性或显性遗传疾病,由COL6A1、COL6A2、COL6A3基因中的一种突变致Ⅵ型胶原蛋白缺陷引起[1]。Ⅵ型胶原蛋白对骨骼肌稳定性、再生和肌细胞基质黏附至关重要。UCMD在我国报道较少。现报道天津市儿童医院2例UCMD患儿临床、肌肉磁共振成像(MRI)及遗传学特点,探讨UCMD的康复管理方案。
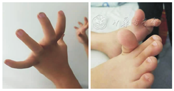
例1,男,4岁,因"至今独走不稳"于2017年10月来诊。患儿系第1胎,第1产,足月儿,因脐带绕颈2周行剖宫产,出生体质量3 kg,出生后颜面部发绀,Apgar评分不详。自幼全身力弱,运动发育落后于同龄儿,5个月翻身,7个月独坐,18个月独走,独走姿势异常,不能自行上楼梯,曾就诊于外院,未确诊。父母非近亲结婚,否认家族遗传史。查体:言语交流可,智力正常。四肢屈侧、双手及足底皮肤柔软,双上肢皮肤散在粟粒样丘疹,下颌一约2.0 cm×0.5 cm瘢痕。高腭弓。四肢肌容积减少。双踝关节挛缩。腕关节、掌指关节、指间关节、跖趾关节、趾间关节活动度大(图1)。小指第一指间关节屈曲挛缩。全身肌力差,闭目力可,鼓腮力稍差,颈曲Ⅳ级,颈伸Ⅴ级,腹肌肌力Ⅱ级,四肢近端肌力Ⅲ级,远端肌力Ⅳ级。双上肢腱反射未引出,双跟腱、膝腱反射(+),双巴氏征(-)。仰卧位坐起困难;不能独自站起;独走呈"鸭步",双足内旋,可见尖足;不能独自上下楼梯;不能跑、跳。实验室检查:肝功能、肾功能、电解质、乳酸均未见异常。肌酸肌酶(CK)102 U/L。四肢神经电生理(2017年10月10日):肌源性损害。头颅MRI、髋关节X线、心电图均未见异常。超声心动图示三尖瓣反流(轻度)。通气功能(2017年12月15日):混合性通气功能中度减低;脉冲震荡肺功能(IOS)检查:外周气道阻力中度增加。上肢皮肤CT检查示皮肤角化。

例2,男,6岁,因"至今不能上楼梯"于2018年6月来诊。患儿系第1胎,第1产,足月因羊水少行剖宫产,出生体质量3.25 kg,Apgar评分不详,出生后哭声弱,力弱,远端关节软。运动发育落后于同龄儿,6个月翻身,8个月会坐,18个月独走但不稳,姿势异常,至今不能独自上楼梯。父母非近亲结婚,否认家族遗传史。查体:言语交流可,智力正常。四肢屈侧、双手及足底皮肤柔软,四肢伸侧可见粟粒样丘疹,额头一约3.0 cm×0.5 cm的瘢痕,下颌一约2.0 cm×0.3 cm的瘢痕。四肢肌容积减少。踝、髋关节挛缩。腕关节、掌指关节、指间关节、跖趾关节,趾间关节活动度大(图2)。全身肌力差,闭目力可,鼓腮力差,颈曲Ⅳ级,颈伸Ⅳ+级,腹肌肌力Ⅱ级,四肢近端肌力Ⅱ~Ⅲ级,远端肌力Ⅲ+~Ⅳ级。双上肢腱反射未引出,双跟、膝腱反射(+),双巴氏征(-)。仰卧位坐起困难;不能独自站起;独走呈"鸭步"双足内旋,右足尖足;不能独自上下楼梯;不能跑、跳。实验室检查:肝功能、肾功能、电解质、乳酸均未见异常。CK为263 U/L。四肢神经电生理(2016年4月20日):肌源性损害。头颅MRI、髋关节X线、心电图均未见异常。超声心动图未见异常。通气功能(2018年6月27日):混合性通气功能轻度减低;IOS检查正常。上肢皮肤CT检查示皮肤角化。
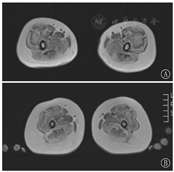
2例患儿下肢肌肉MRI T1加权像(T1WI)均示:大腿肌肉弥散脂肪化,单块肌肉受累边缘重中央轻,以股外侧肌明显,呈"三明治"征,股直肌中央亦有受累,缝匠肌、长收肌、股薄肌相对受累较轻。小腿水平弥散受累,以腓肠肌和比目鱼肌外侧受累为著(图3)。
对患儿基因组DNA进行全外显子组捕获和测序,并对候选变异位点在家系样本DNA中进行Sanger测序验证。
例1:COL6A3基因c.6210+1G>C杂合突变(图4),为新发变异。该突变位于内含子和外显子边界区域即供体点,理论上导致外显子跳跃变异,曾见于1例肌营养不良患者的体细胞嵌合变异。经验证,其父母该位点为野生型(图4)。
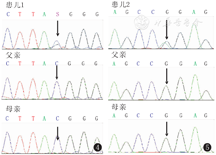
例2:COL6A1基因Exon9 c.850G>A (p.Gly284Arg)杂合突变(见图5),为错义突变,曾在UCMD患者中检测到该变异[2] 。经验证,其父母该位点均为野生型(图5)。
本研究经医院医学伦理委员会批准,患儿监护人均知情同意。
UCMD于1930首次被Ullrich描述为"先天性硬化性肌营养不良"。主要临床特点为早发型全身肌无力、远端关节活动度大、近端关节挛缩,但智力正常[3]。UCMD患儿通常不能独立行走或仅能短时间独立行走[4]。其他特征包括:皮肤过度角化;四肢伸面、手掌和脚底皮肤柔软;伤口愈合异常导致瘢痕或"香烟纸"样瘢痕形成;跟骨突出;面部肌肉受累和高腭弓;先天性髋关节脱位;生后短暂性后凸畸形[4]。随疾病进展,可出现脊柱僵硬、脊柱侧凸及近端关节挛缩;第一指间关节屈曲挛缩和跟腱紧张;呼吸肌受累较早,如果不接受夜间呼吸支持,在第1或第2个十年,常死于呼吸衰竭;心脏无明显受累;血清CK常正常或轻度升高[4]。本研究2例患儿临床特点与UCMD临床特点相符。
本研究2例患儿肌肉MRI病变模式与UCMD患者下肢MRI研究一致[5] 。因MRI结果需高水平专家解释,故在神经肌肉疾病诊断中不作为常规基础检查,对特定患者,可作为鉴别诊断附加工具。
UCMD主要与Bethlem肌病相鉴别,二者为等位基因病。Bethlem肌病病情相对轻,进展缓慢,成人期仍有移动能力,呼吸肌受累不显著,寿命较少受影响[6],下肢肌肉MRI可见受累肌肉周边信号增高而中央相对正常,股外侧肌和股中间肌有同心脂肪浸润,股直肌可见"中心影"征或"U"形浸润[6]。本研究2例患儿起病早,呼吸肌受累早,肌肉MRI均可见股外侧肌呈"三明治"征,故目前临床支持考虑UCMD。
本研究例1患儿COL6A3基因c.6210+1G>C杂合突变为新发变异,结合患儿临床特征和肌肉MRI特点,考虑此新发变异可致UCMD,扩展了COL6A3基因突变内容。例2患儿COL6A1基因Exon9 c.850G>A (p.Gly284Arg)杂合突变,为错义变异,其临床特点和肌肉MRI特点亦符合UCMD,与既往文献报道一致[2]。
UCMD发病机制尚不明确。研究表明,其与线粒体功能障碍有关[7,8]。线粒体渗透性转换孔(PTP)的开放是决定细胞凋亡和坏死的重要因素。研究提示PTP开放在所有Ⅵ型胶原肌病中起关键作用,为患者药物治疗开辟了新前景[9]。环孢素(CsA)可抑制PTP开放[7]。Merlini等[10]研究表明,中剂量CsA[5 mg/(kg·d)]治疗对Ⅵ型胶原肌病的线粒体功能有利,并可显著降低患者肌细胞死亡频率,给Ⅵ型胶原肌病的治疗带来了巨大希望。
UCMD患儿管理重点是普通医疗支持及预防和治疗并发症[11]。减少废用性肌无力,延缓肌无力进展;防治脊柱侧弯及关节挛缩;合理选择和使用支具、辅助器具;改善肺功能,加强气道管理,积极预防和治疗肺部感染;出现低通气时,辅助通气支持治疗;注意营养管理,改善吞咽,经口进食困难无法维持体质量及营养需求时,建议安置胃管;注意安全防护和监测。
所有作者均声明不存在利益冲突

























