
提高对软骨发育不全(ACH)临床表型及基因型的认识。
回顾性分析2例ACH患儿的临床资料及基因检测结果,并复习相关文献。
例1,女,1岁,母亲身材矮小,因发现双下肢膝反张9月余入院。查体:头围45 cm,身材矮小,四肢较短,双下肢肌张力偏低,发育商65分,X线示双侧髂骨及双髋关节病变,考虑ACH,送检基因结果示FGFR3基因c.1138G>A杂合变异(p.Gly380Arg),母亲该基因位点为杂合变异。例2,女,10个月,因发现四肢无力5月余入院。查体:头围46 cm,身材矮小,四肢短小,四肢肌张力减低,四肢肌力4级,发育高41分,X线示双下肢符合ACH。送检基因结果示FGFR3基因c.1138G>A杂合变异(p.Gly380Arg)为新生变异,父母该基因位点为野生型。
ACH临床特征具有多样性,骨骼改变和神经发育同样需要加强认识和甄别。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
遗传性侏儒主要包括软骨发育不全(achondroplasia,ACH)和软骨发育不良(hypochondroplasia,HCH),ACH最多见,为软骨化骨缺陷而膜化骨正常的一种发育异常[1]。二者临床特征均为身材矮小,典型临床表现为四肢过短、巨头、前额突出、椎骨茎缩短,均呈常染色体显性遗传,为成纤维细胞生长因子受体3(FGFR3)基因变异所致,因85%的患者双亲表型正常,故多为新生变异所致。HCH症状较ACH轻,二者因表型差异细微而不易区分。研究显示FGFR3基因变异与表型关系确切,98%的ACH患者由FGFR3基因Gly380Arg变异引起,而60%~65%的HCH患者由FGFR3基因Asn540Lys变异所致,且前者具有特殊父龄效应,即父亲生育年龄超过35岁时,其后代患病风险显著增加[2]。ACH的发病率为1/26 000~1/16 000活产儿,患儿智力常正常。罹患ACH的胎儿常在胎龄25周后四肢长骨生长出现明显落后,确诊时多数已至妊娠晚期。因超声、产科及儿科医师对本病认识不足造成妊娠期及新生儿期不能早期确诊。本研究回顾性分析2例均合并脑积水并伴全面发育迟滞的ACH患儿临床资料及基因检测结果,以期加强对本病的认识。
例1,女,1岁,因发现双下肢膝反张9月余入院。9月余前患儿双膝反张,肌张力低下,头大,不能抬头及翻身,于2019年7月15日来诊,行髋关节正位及蛙式位X线平片检查考虑ACH,后转至北京某医院检查后建议康复训练,8月26日再次来诊并入院治疗。患儿系第1胎,第1产,胎龄34+2周行剖宫产出生,出生时有窒息抢救史。父亲左上肢瘫痪,但可行走;母亲身高130 cm,非近亲结婚。入院查体:发育不良,抱入病房,身材矮小,头围45 cm,双下肢肌张力低,四肢肌力低下,四肢短小。专科检查:仰卧位,头可居于中线,追视追听欠灵活,双手可居中线位活动,双下肢屈曲;俯卧位,被动肘支撑,可瞬间抬头;坐位,全前倾坐;立位,双下肢可支撑片刻;手抓位,无主动取物意识。异常姿势:双下肢外旋外展;肌张力:双下肢稍偏低;关节活动度:股角180°,腘窝角170°,足背屈角30°,围巾征不过中线,跟耳征(+);语言及智力评价:可逗笑,偶尔可笑出声,蒙面试验不可引出,发育商65分。辅助检查:头颅磁共振成像(MRI)示轻度幕上脑积水;髋关节正位及蛙式位X线片考虑ACH。入院诊断:(1)遗传代谢病?(2)全面发育迟滞;(3)ACH;(4)脑积水。入院后进一步检查:甲状腺功能、维生素D未见异常。予电子生物反馈疗法、中频脉冲电治疗、慢性小脑电刺激术、关节松动训练等理疗及康复运动训练。4周后病情相对好转,患儿家属要求出院。出院后间断康复训练,现会独坐、会爬。
患儿及父母血标本送至北京迈基诺医学检验所行基因测序分析发现,FGFR3基因第9号外显子上c.1138G>A核苷酸变异,致第380号氨基酸甘氨酸变异为精氨酸,为错义变异,其致病性已有文献报道(参考数据库:HGMD Pro及PubMed)。该变异不属于多态性变化,在人群中发生的频率极低(参考数据库:1000Genomes、dbSNP)。家系验证发现患儿父亲该位点为野生型,母亲该位点为杂合变异(图1)。
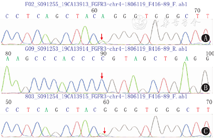
根据美国医学遗传学与基因组学学会(ACMG)联合美国分子病理学协会(AMP)在2015年的"基因序列变异的解释标准和指南"行致病性分析[3],FGFR3基因c.1138G>A变异证据强度为"PS1+PM2+ PP3",为疑似致病性变异。结合患儿临床表现及基因检测结果诊断ACH明确。
例2,女,10个月,因发现四肢无力5月余于2019年3月入院。5月余前发现患儿四肢无力,表现为四肢活动少,活动范围增大,伴竖头不稳及反应迟钝,曾就诊于外院,头颅MRI提示脑积水,予康复治疗2个疗程好转出院。患儿系第3胎,第3产,足月顺产出生,出生时无窒息抢救史。父母及两哥哥均体健,父母非近亲结婚。入院体格检查:发育不良,抱入病房,身材矮小,头围46 cm,鼻梁塌陷,前额部突出,四肢肌张力减低,四肢肌力4级,四肢短小。专科检查:仰卧位,头可居于中线,追视追听可,双手可居中线位活动,双下肢屈曲,非对称性紧张性颈反射(asymmetrical tonic neck reflex,ATNR)姿势消失;俯卧位,辅助下可双手支撑,可从仰卧位翻身至俯卧位;坐位,半前倾位;立位,双下肢不可支撑;手抓位,双手可全手掌抓物;肌张力,四肢肌张力减低;股角180°,腘窝角150°,足背屈角50°,围巾征过中线,跟耳征(+)。语言及智力评价:可逗笑,笑出声,会无意识喊"mama",呼名反应差,不会玩躲猫猫游戏,发育商41分。小儿双下肢正位X线片考虑ACH。入院后诊断:(1)ACH;(2)全面发育迟滞;(3)脑积水。予关节松动、理疗等治疗25 d后好转出院。
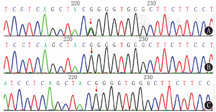
基因基因检测:患儿及父母血标本送至北京康旭医学检验有限公司行基因测序发现,FGFR3基因c.1138G>A杂合核苷酸变异,该变异导致第380号氨基酸由Gly变为Arg(p.Gly380Arg),为错义变异,可导致蛋白质功能受影响。该变异致病性已有文献报道(参考数据库:HGMD Pro及PubMed)。该变异不属于多态性变化,在人群中发生的频率极低(参考数据库:1000Genomes、dbSNP)。家系验证显示父母FGFR3基因均未发现上述变异,为新生变异(图2)。
本研究通过医院医学伦理委员会批准(批准文号:2020191),患儿监护人均知情同意。
FGFR3基因定位于4p16.3,全长约16.5 kb,包含19个外显子和18个内含子,编码806个氨基酸残基组成的FGFR3。FGFR3受体蛋白包括3个部分:(1)胞外区为3个Ig样的配体结合区(IgⅠ、IgⅡ和IgⅢ);(2)跨膜区;(3)胞内区为酪氨酸激酶区(TK1和TK2)。就目前所知,几乎所有ACH患者都是由于FGFR3的跨膜区变异引起,其中98%为G380R变异,尚有G375C或G346E变异。FGFR3为跨膜酪氨酸激酶受体家族,多种成纤维细胞生长因子(FGF)可作为其配体,在软骨发育与软骨稳态维持中发挥作用,FGF通过类肝素硫酸蛋白聚糖作用与FGFR结合致FGFR二聚化而活化。
FGF信号在骨形成和发育过程中的作用至关重要。FGFs家族由22种配体和4种受体组成,分别是FGFR1~FGFR4,它们之间在氨基酸水平上有同源性。FGFs通过结合FGFRs发挥作用。FGFRs在骨形成过程中有特异的时空表达模式。FGFR3在骨发育早期先于间充质凝集中心软骨细胞中表达,随后在生长板软骨和关节软骨增殖带和肥大前带软骨细胞中表达。FGFR3为骨骼生长的负性调控分子,FGFR3的10多种功能增强型变异可致多种人类软骨发育障碍性侏儒,如ACH和HCH等。变异后的FGFR3功能持续激活和调控,不依赖成纤维细胞生长因子,导致FGFR3对骨骼生长的负向调节作用失控造成患者骨骼发育和生长障碍。FGFR3基因变异导致蛋白质结构改变而造成功能改变致ACH的发生。95%以上ACH为FGFR3功能增强型变异所致。而FGFR3敲除小鼠表现骨骼过度生长、骨量减少及骨骼矿化障碍,支持FGFR3对骨骼生长的负向调节作用。
已报道ACH和HCH致病FGFR3基因存在变异热点,即Gly380Arg和Asn540Lys[4]。本组2例患儿均为FGFR3基因Gly380Arg杂合变异,例2为新生变异,支持文献报道。因ACH多为新生变异引起,但几乎所有新生变异均发生于父方的等位基因,提示FGFR3在精子发生时变异性增强和在雄性细胞系中FGFR3存在该致病变异的选择性优势[5]。
有报道人生长激素对本病的治疗有一定疗效,能改善患儿身高,且无严重不良反应[6]。但本研究中2例ACH患儿因费用问题未使用该药。
ACH患儿智力多正常,Priestley和Lorber[7]报道伴头颅畸形的ACH患儿10例,其中1例罹患进行性脑积水,合并智力低下及痉挛性四肢瘫,本研究2例患儿均伴有脑积水和全面发育迟滞,考虑患儿头围较大,ACH可能影响颅骨发育进而影响脑发育致全面发育落后。而Wynne-Davies和Patton[8]报道有10%~20%的HCH患儿合并智力低下。ACH和HCH临床表型不易区分,故例2患儿可能为HCH,但基因型更支持ACH,推测全面发育迟滞与继发性脑积水有关。
因本病目前无特殊治疗方法,故产前诊断尤为重要,对彩超发现肢体短缩胎儿结合遗传学筛查可明确诊断,且可早期诊断,同时减轻孕妇及家属焦虑。
所有作者均声明不存在利益冲突

























