
回顾分析深圳市儿童医院儿童重症医学科(PICU)收治的1例迟发型戊二酸血症Ⅱ型患儿的临床资料和治疗经过。患儿,女,以进行性近端肌无力加重为主要表现,伴呕吐、腹痛、肝大,血清转氨酶、胆红素和肌酸激酶增高,高血氨,低血糖,代谢性酸中毒。血液氨基酸及酰基肉碱谱分析见多种酰基肉碱升高,肌肉活检为脂质沉积性肌病,全外显子组测序显示电子转运黄素蛋白脱氢酶(ETFDH)基因纯合突变。经过维生素B2、左卡尼汀、辅酶Q10及血浆置换治疗,患儿肌力恢复,各项生化指标恢复正常。对临床上出现迅速进展的肌病,伴肝损害、血清肌酸激酶增高的患儿,应警惕迟发型戊二酸血症Ⅱ型。提高认识是早期诊断和治疗的关键。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
戊二酸血症Ⅱ型(GA Ⅱ)是一种罕见的常染色体隐性遗传脂肪酸氧化代谢障碍性疾病,可分为3型:新生儿期发病伴先天畸形、新生儿期发病无先天畸形、轻型或迟发型[1]。迟发型患儿可在各个年龄起病,临床表现多样且无特异性。轻者无症状,重者可出现呼吸衰竭[2,3]。目前为止国内报道较少,个体差异大,可累及多脏器,容易误诊。现回顾性分析深圳市儿童医院收治的1例迟发型GA Ⅱ患儿的临床资料,以提高临床医师对本病的认识。
患儿,女,13岁8个月,因"乏力伴消瘦1个月,发现肝功能异常10 d"入院。患儿入院1个月前无明显诱因出现乏力,体力劳动后明显,休息可减轻,进行性加重,伴体质量进行性降低,从37 kg降至31.8 kg,食欲差。病程中有间断呕吐,进食后明显,伴腹痛,腹上区痛为主。起病0.5个月后至外院就诊,血清丙氨酸转氨酶(ALT) 66 IU/L,天冬氨酸转氨酶(AST) 209 IU/L,乳酸脱氢酶(LDH) 1 467 U/L,肌酸激酶(CK) 780 U/L。胃镜检查提示胆汁反流性糜烂性胃炎。予"还原型谷胱甘肽、奥美拉唑、营养支持等"治疗无效,复查血清ALT 262 U/L,AST 551 U/L,LDH 2 616 U/L,CK 2 484 U/L,转至深圳市儿童医院。
既往患儿体健,无肝炎患者密切接触史,无输血史,否认家族中有类似症状患者及遗传病史。父母非近亲婚配,第1胎,第1产,足月顺产,否认出生时抢救窒息史,智力和运动发育与同龄儿相仿,按计划疫苗接种。
入院体格检查:体温36.0 ℃,心率88次/min,呼吸20次/min,血压121/88 mmHg(1 mmHg=0.133 kPa),经皮血氧饱和度98%,体型消瘦,神志清楚,全身皮肤及黏膜无黄染,无肝掌及蜘蛛痣,呼吸平顺,节律规则,双肺呼吸音清,未闻及啰音;心音有力,律齐,未闻及杂音;肝右肋下平脐,质韧,触痛,四肢及躯干感觉正常,腓肠肌压痛明显,四肢肌力4级,肌张力正常。
入院后予还原型谷胱甘肽、奥美拉唑静脉输注等治疗。同时完善相关检查。患儿肌力仍进行性减低,四肢远端肌力正常,近端肌力减低,1级,双侧膝反射减弱。住院第3天患儿出现呼吸困难,胸闷明显,吞咽及咳嗽反射弱,转儿童重症医学科(PICU)监护治疗,行气管插管机械通气。复查血清ALT 377 U/L,AST 3 601 U/L,LDH 7 230 U/L,CK 15 353 U/L,肌红蛋白3 254.0 μg/L,血氨80.9 μmol/L,乳酸7.29 mmol/L,总胆红素68.6 μmol/L,结合胆红素40.1 μmol/L,血糖1.9 mmol/L。行血浆置换治疗共5 d。液相串联质谱法血液氨基酸及酰基肉碱谱分析提示短、中及长链酰基肉碱升高,肉豆蔻烯酰肉碱0.406 μmol/L(参考值0~0.22 μmol/L),肉豆蔻二烯酰肉碱0.089 μmol/L(参考值0~0.05 μmol/L),棕榈烯酰肉碱0.639 μmol/L(参考值0.02~0.39 μmol/L)。尿有机酸分析未见异常。血/尿淀粉酶、尿铜正常范围。住院期间2次脑脊液(入院第3天及入院2周)常规、生化检查及培养未见异常。血液自身免疫性周围神经病检查、乙酰胆碱受体抗体及自身抗体谱未见异常。
腹部超声显示肝肋下平脐,肝实质回声增强、增粗、欠均。腹上区CT平扫+增强扫描提示肝脏密度弥漫减低,CT值约-45 HU(亨氏单位)。颅脑及脊柱磁共振(MR)平扫增强及股四头肌MR未提示明显异常。双下肢肌电图提示各肌肉运动单位电位(MUP)波幅高、相位增多、时程延长,提示肌肉病损,近端重。
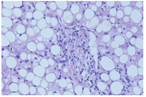
右股外侧肌肌肉活检提示轻度肌损害改变,考虑脂质沉积性肌病(图1)。肝活检提示肝细胞脂肪变性(图2)。
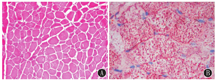
注:ORO:油红O ORO:oil-Red-O
基因检测采用全外显子二代测序,一代验证(北京金准医学检验所用),患儿ETFDH基因存在一处纯合突变c.250G>A(p.A84T),提示GAⅡ。家系验证证实父母均为电子转运黄素蛋白脱氢酶(ETFDH)基因c.250G>A杂合突变携带者。加用维生素B1(30 mg,2次/d)及B2(15 mg,2次/d)、左卡尼汀(2 g,1次/d)、辅酶Q10(10 mg,3次/d)口服,治疗后患儿肝功能好转,血清转氨酶、CK、乳酸、血糖等生化指标逐渐恢复正常,咳嗽反射、吞咽反射逐渐恢复,四肢肌力恢复至V级,腱反射可引出。机械通气23 d,PICU住院治疗1个月后转入普通病房。继续予口服维生素B2、左卡尼汀、辅酶Q10支持治疗,配合康复训练。1周后带药出院,出院时体质量31 kg。出院3个月后随访,患儿规律口服维生素B2(20 mg,3次/d),未再出现乏力等不适症状。本研究通过深圳市儿童医院医学伦理委员会批准[批准文号:深儿医伦审(文章)2019(015)号],患儿监护人均知情同意,并签署知情同意书。
GA Ⅱ是由于编码线粒体的电子转运黄素蛋白(ETF)α或β亚单位和ETFDH的基因先天缺陷或突变引起的多种酰基辅酶A脱氢酶缺乏,导致线粒体内脂肪酸β氧化受阻,能量供应障碍,中间代谢产物积累,引起肌病、肝病或多器官功能损害。Przyrembel等[4]在1976年首次报道本病。迟发型GA Ⅱ临床表现复杂多样。年长儿及成人患儿常起病隐匿,早期症状无特异性,常以肌无力、肌肉痛、运动耐力下降、脂肪肝、低血糖、肝功能异常、血清肌酸激酶不同程度升高,急性期可伴代谢性酸中毒、低酮性低血糖、高脂血症等代谢紊乱。患儿常合并脂肪肝、脂质沉积性肌病等[2,3,5],容易误诊为多发性肌炎、吉兰-巴雷综合征、肝炎等疾病[6,7]。
本例患儿病初疑诊消化道疾病,治疗无效,后出现进行性近端肌无力和肝损害,伴呼吸困难,曾考虑为吉兰-巴雷综合征。患儿血清转氨酶、肌酸激酶短时间内迅速增高,伴低血糖、代谢性酸中毒、血氨及胆红素增高,考虑肝衰竭。梁雁等[1]报道了1例12岁女性患者,病初表现为肌无力,曾误诊为"脑白质病",予泼尼松治疗数年,停药后肌无力加重,伴呕吐、意识障碍,最终经尿有机酸分析诊断。吴娜等[2]报道1例32岁男性患者,反复乏力,全身酸痛,曾误诊为"横纹肌溶解",予血液透析后仍有乏力,最终经肌活检病理诊断。崔亚杰等[5]报道1例11岁男性患儿,双下肢肌无力,意识障碍,伴肌肉疼痛,曾疑诊"重症肌无力、多发性肌炎",予泼尼松、复合B族维生素治疗,症状改善,停药后反复,最终通过基因分析诊断。杨艳玲等[8]报道了3例患儿曾疑诊为"心肌炎、多发性肌炎、皮肌炎、周期性麻痹、肝炎"等疾病,接受激素、丙种球蛋白、保肝等治疗无效。
GA Ⅱ确诊需要借助实验室检查:(1)血液氨基酸及酰基肉碱谱分析和尿有机酸分析。典型患儿血液多种短、中和长链酰基肉碱升高,尿液戊二酸、乙基丙二酸、辛二酸等多种有机酸升高[2,3]。(2)肌肉病理检查,GA Ⅱ患儿常有肌肉脂质沉积,脂质沉积性肌病是本症的病理特征之一。典型患儿肌纤维内大量脂滴沉积,以Ⅰ型肌纤维受累为主,改良Gomori三原色染色可见破碎肌红纤维,电镜下可见脂质沉积性肌病的病理改变[9]。(3)基因检测:ETF的α和β亚单位分别由ETFA和ETFB基因编码,ETFDH由ETFDH基因编码。从GA Ⅱ患儿受累基因概率上看,ETFA、ETFB、ETFDH的基因突变频率分别为27%、27%和47%,提示ETFDH缺陷突变在迟发型GA Ⅱ中最常见[10,11]。我国南、北方人群存在突变差异。南方人群以c.250G>A为高频突变热点[12],北方人群以c.770A>G和c.1227A>C为突变热点[9]。
迟发型GA Ⅱ是可以治疗的遗传代谢病,对不明原因的肌肉病、肝病、代谢紊乱患儿,应及早进行血液氨基酸及酰基肉碱谱分析、尿有机酸分析、肌活检及ETFA、ETFB和ETFDH基因突变检测。值得注意的是,本例患儿未检测出有机酸尿症,提示迟发型GA Ⅱ患儿可能仅在病情加重时出现有机酸尿症,而在发作间期生化代谢分析结果可能正常[2]。本例患儿血液多种酰基肉碱升高,肌活检提示脂质沉积性肌病,最后通过ETFDH基因检测诊断迟发型GA Ⅱ。
大剂量维生素B2可改善患儿临床症状,早期治疗能够改善预后[1]。对伴辅酶Q10缺乏的迟发型GA Ⅱ患儿,辅酶Q10与维生素B2联用可改善症状。可静脉输注葡萄糖,予左卡尼汀和甘氨酸,促进有毒酸性代谢物质排出,尽快纠正代谢性酸中毒[2]。本例患儿因为肝衰竭进行了血浆置换治疗,同时补充维生素B2、左卡尼汀及辅酶Q10。患儿肌力恢复,肝功能等各项生化指标恢复正常,带药出院。
综上,迟发型GA Ⅱ临床表现缺乏特异性,早期容易被误诊。对临床上迅速进展、以近端肌无力为特点的肌病,尤其伴随肝损害、血清CK增高、高血氨、低血糖、代谢性酸中毒等代谢异常的患儿,应高度怀疑本病,并尽早进行血液氨基酸及酰基肉碱谱分析、尿有机酸分析、肌活检和基因检测,争取早期明确诊断,给予补充维生素B2等治疗。
所有作者均声明不存在利益冲突

























