分析17α-羟化酶/17,20碳链裂解酶缺陷症(17OHD)的临床及CYP17A1基因突变特征。
收集2014年3月至2019年12月首都儿科研究所附属儿童医院内分泌科收治的6例17OHD患儿的临床资料、实验室检查结果、CYP17A1基因突变情况,统计同期所有先天性肾上腺皮质增生症(CAH)的类型及数量。总结17OHD的临床特征及发病比例。
6例患儿来自5个家庭,年龄1岁6个月~15岁,其中46,XX 2例,46,XY 4例;社会性别均为女性。临床表现中高血压3例(50.0%),低血钾4例(66.7%),阴唇包块1例(16.7%)。46,XY者性腺发育为睾丸;46,XX者子宫卵巢发育不良。实验室检查:8 AM皮质醇降低,为0.62~5.93 mg/L;促皮质素(ACTH)升高5例,为84~271 ng/L,1例ACTH正常(58 ng/L)的患儿行ACTH激发试验后皮质醇峰值仅为1.75 mg/L;6例均孕酮升高而17羟孕酮低下;睾酮、雌二醇减低,黄体生成素(LH)、卵泡刺激素(FSH)升高。肾上腺CT均示轻度增生。在114例同期诊断CAH患儿中,17OHD占5.3%,发病率居第2位。CYP17A1基因突变分析显示2例患儿p.Y329fs (c.985_987delTACinsAA)纯合突变;2例同胞患儿为p.Y329fs及外显子1-7缺失的复合杂合突变;1例患儿为p.Y329fs及p.R416C(c.1246C>T)的复合杂合突变;1例患儿p.L465P (c.1394T>C)纯合突变。p.L465P (c.1394T>C)首次见于中国人群。
在CAH中,17OHD并不少见,对于女性外观的儿童低钾血症、高血压及性腺功能低下者警惕17OHD存在。p.Y329fs为中国儿童17OHD热点突变;p.L465P (c.1394T>C)为中国人群新发突变,其丰富了中国人CYP17A1基因突变谱系。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
17α-羟化酶/17,20-碳链裂解酶缺陷症(17OHD)是一种罕见的先天性肾上腺皮质增生症(CAH),呈常染色体隐性遗传,其典型的临床症状包括高血压、低血钾、第二性征不发育、女性原发性闭经、男性假两性畸形等。儿期早期症状相对较轻,儿科医师对本病认识不足,易延误诊治。有关儿童17OHD,多以病例报告为主。本研究报道6例17OHD患儿,总结其临床特点,分析其基因突变类型,以加深临床医师对本病的认识。
回顾性分析2014年3月至2019年12月首都儿科研究所附属儿童医院内分泌科收治的6例基因检测明确诊断的17OHD患儿的临床资料及基因检测结果。统计同期收治入院的所有类型新发CAH患儿。本研究通过首都儿科研究所附属儿童医院伦理委员会批准(批准文号:SHERLL2018005),患儿监护人知情同意并签署知情同意书。
收集17OHD患儿首诊及末次随访的基本资料,包括:(1)起病年龄、性别、主诉;(2)身高、肤色、血压、Tanner分期;(3)血气分析、电解质、类固醇激素水平、肾素醛固酮水平、染色体分析;(4)左腕正位及肾上腺和性腺影像;(5)收集同期其他明确诊断的CAH患儿类型。
电化学发光法检测皮质醇、促皮质素(ACTH)及性激素;化学发光法检测17羟孕酮(17-OHP)及肾素、醛固酮;酶联免疫法测定脱氢表雄酮(DHEA);培养法G显带行染色体核型分析;骨龄分析根据社会性别采用G-P图谱法。
留取先证者及其父母静脉血各3 mL,提取DNA,用聚合酶链反应(PCR)进行CYP17A1基因扩增,用ABI3730xl测序仪(美国Applied Biosystems公司)以Sanger测序法进行测序,测序结果使用"Mutation Surveyor"软件与参考序列进行比对分析。与父母来源不符合者行多重连接探针扩增技术(MLPA)检测待检样本基因外显子缺失/重复突变。
6例患儿来自5个家庭,临床资料见表1。年龄1岁6个月~15岁,其中46,XX 2例,46,XY 4例;社会性别均为女性。临床表现中,本组患儿肤色均正常,均无色素沉着,高血压3例(50.0%),低血钾4例(66.7%),阴唇包块1例(16.7%)。血压最高达190/145 mmHg(1 mmHg=0.133 kPa),并高眼压症(例6),余患儿无高血压脏器损害征象;患儿低血钾轻重不一,重者于呼吸道感染后出现呼吸肌麻痹、呼吸衰竭,予呼吸机支持治疗(例5),轻者肺炎后血钾轻度降低(例1),2例虽有严重高血压,但血钾正常(例3、例6)。6例患儿均女性外阴,其中4例46,XY患儿均未见阴道口,无阴蒂肥大或阴茎,内生殖器与染色体核型一致,可见睾丸组织位于大阴唇下、腹股沟或膀胱外上方,无卵巢组织;2例46,XX者卵巢及子宫发育不良。1例乳房发育良好,B4期,未见腋毛,可见少许绒毛状阴毛,P2期(例6),其余患儿性发育均为Tanner Ⅰ期。
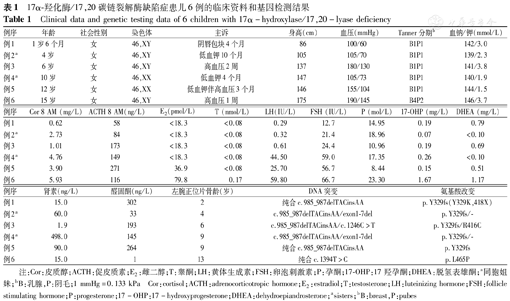
17α-羟化酶/17,20碳链裂解酶缺陷症患儿6例的临床资料和基因检测结果
Clinical data and genetic testing data of 6 children with 17α-hydroxylase/17,20-lyase deficiency
17α-羟化酶/17,20碳链裂解酶缺陷症患儿6例的临床资料和基因检测结果
Clinical data and genetic testing data of 6 children with 17α-hydroxylase/17,20-lyase deficiency
| 例序 | 年龄 | 社会性别 | 染色体 | 主诉 | 身高(cm) | 血压(mmHg) | Tanner分期b | 血钠/钾(mmol/L) |
|---|---|---|---|---|---|---|---|---|
| 例1 | 1岁6个月 | 女 | 46,XY | 阴唇包块4个月 | 86 | 100/60 | B1P1 | 142/3.0 |
| 例2a | 4岁 | 女 | 46,XY | 低血钾10个月 | 105 | 105/70 | B1P1 | 139/2.3 |
| 例3 | 6岁 | 女 | 46,XY | 高血压2周 | 137 | 180/130 | B1P1 | 141/3.8 |
| 例4a | 10岁 | 女 | 46,XX | 低血钾4个月 | 147 | 105/73 | B1P1 | 140/1.9 |
| 例5 | 12岁 | 女 | 46,XX | 低血钾伴高血压3个月 | 146 | 155/104 | B1P1 | 144/1.5 |
| 例6 | 15岁 | 女 | 46,XY | 高血压1周 | 175 | 190/145 | B4P2 | 146/3.7 |
| 例序 | Cor 8 AM (mg/L) | ACTH 8 AM(ng/L) | E2(pmol/L) | T (nmol/L) | LH(IU/L) | FSH (IU/L) | P (mol/L) | 17-OHP (mg/L) | DHEA (mg/L) |
|---|---|---|---|---|---|---|---|---|---|
| 例1 | 0.62 | 58 | <18.3 | <0.08 | 0.29 | 12.7 | 14.95 | 0.19 | 0.79 |
| 例2a | 2.73 | 84 | <18.3 | <0.08 | 0.32 | 21.4 | 18.96 | 0.07 | <0.10 |
| 例3 | 1.01 | 173 | <18.3 | <0.08 | 0.61 | 24.4 | 10.96 | 0.19 | 0.69 |
| 例4a | 4.76 | 149 | <18.3 | <0.08 | 44.50 | 59.0 | 17.35 | 0.26 | <0.10 |
| 例5 | 3.90 | 271 | 36.9 | <0.08 | 25.70 | 56.7 | 8.44 | 0.15 | 0.51 |
| 例6 | 5.93 | 116 | 79.8 | 0.17 | 59.80 | 66.7 | 23.30 | 1.67 | 1.17 |
| 例序 | 肾素(ng/L) | 醛固酮(ng/L) | 左腕正位片骨龄(岁) | DNA突变 | 氨基酸改变 |
|---|---|---|---|---|---|
| 例1 | 15.0 | 302 | 2 | 纯合c.985_987delTACinsAA | p.Y329fs(Y329K,418X) |
| 例2a | 60.0 | 33 | 4 | c.985_987delTACinsAA/exon1-7del | p.Y329fs/- |
| 例3 | 1.9 | 193 | 6 | c.985_987delTACinsAA/c.1246C>T | p.Y329fs/R416C |
| 例4a | 498.0 | 145 | 9 | c.985_987delTACinsAA/exon1-7del | p.Y329fs/- |
| 例5 | 90.0 | 264 | 9 | 纯合c.985_987delTACinsAA | p.Y329fs |
| 例6 | 15.0 | 1 | 13 | 纯合c.1394T>C | p.L465P |
注:Cor:皮质醇;ACTH:促皮质素;E2:雌二醇;T:睾酮;LH:黄体生成素;FSH:卵泡刺激素;P:孕酮;17-OHP:17羟孕酮;DHEA:脱氢表雄酮;a同胞姐妹;bB:乳腺,P:阴毛;1 mmHg=0.133 kPa Cor:cortisol;ACTH:adrenocorticotropic hormone;E2:estradiol;T:testosterone;LH:luteinizing hormone;FSH:follicle stimulating hormone;P:progesterone;17-OHP:17-hydroxyprogesterone;DHEA:dehydroepiandrosterone;asisters;bB:breast,P:pubes
结果见表1。6例患儿8 AM皮质醇水平均降低,5例患儿ACTH升高。1例行ACTH激发试验后皮质醇由0.62 mg/L升至1.75 mg/L(例1)。所有患儿睾酮及雌二醇水平低下,黄体生成素(LH)及卵泡刺激素(FSH)明显升高;孕酮水平明显升高;17-OHP及DHEA低下。肾素水平正常或低下3例,醛固酮水平低下2例。肾上腺增强CT可见双侧或单侧肾上腺轻度增大,肾上腺影像改变与临床低血钾或高血压程度无关。左腕正位片中,4例年龄≤10岁的患儿骨龄与年龄接近(例1~4),年长者(年龄>10岁)骨龄落后于年龄(例5~6),所有患儿骨龄身高大于同龄健康女童。
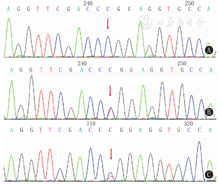
结果见表1。8号外显子c.1394T>C(p.L465P)在中国人中首次报道(图1);6号外显子c.985_987delTACinsAA(p.Y329fs)最常见,占等位基因的58.3%(7/12个)。除外显子1-7的大片段缺失突变外,其余突变均位于外显子6或8。患儿父母均为携带者,无临床症状。所有突变符合美国医学遗传学与基因组学学会(ACMG)指南的致病性突变。
例1~5处于生长发育中,予对身高增长影响最低的氢化可的松治疗,约10 mg/m2,分3次口服;例6予泼尼松治疗,5 mg/d,分2次口服。加药后所有患儿血钾正常,高血压者血压好转,后加用盐皮质激素受体拮抗剂螺内酯后,血压可降至正常。为评价睾丸功能及选择睾丸摘除手术时机,例1在2.5岁进行了人绒毛膜促性腺素(HCG)试验,试验后睾酮仍测不出(<0.08 nmol/L)。例5现随访5年,最初血压控制良好,14岁开始口服雌激素替代治疗。患儿近2年用药不规律,17岁复诊血压升高,乳房B2期,无阴毛,间断血钾降低及肾脏损害(微量白蛋白尿),复查盆腔超声仍显示子宫幼稚,卵巢小,最大卵泡2 mm,双能X线(DXA)检查提示全身骨密度严重降低;末次随访结果见表2。
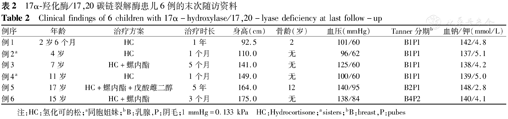
17α-羟化酶/17,20碳链裂解酶患儿6例的末次随访资料
Clinical findings of 6 children with 17α-hydroxylase/17,20-lyase deficiency at last follow-up
17α-羟化酶/17,20碳链裂解酶患儿6例的末次随访资料
Clinical findings of 6 children with 17α-hydroxylase/17,20-lyase deficiency at last follow-up
| 例序 | 年龄 | 治疗方案 | 治疗时长 | 身高(cm) | 骨龄(岁) | 血压(mmHg) | Tanner分期b | 血钠/钾(mmol/L) |
|---|---|---|---|---|---|---|---|---|
| 例1 | 2岁6个月 | HC | 1年 | 92.5 | 2 | 101/60 | B1P1 | 142/4.8 |
| 例2a | 4岁 | HC | 1个月 | 110.0 | 无 | 96/62 | B1P1 | 137/5.1 |
| 例3 | 7岁 | HC+螺内酯 | 5个月 | 141.0 | 无 | 125/60 | B1P1 | 138/4.2 |
| 例4a | 11岁 | HC | 1个月 | 149.0 | 无 | 100/60 | B1P1 | 139/5.0 |
| 例5 | 17岁 | HC+螺内酯+戊酸雌二醇 | 5年 | 164.0 | 12 | 140/95 | B2P1 | 148/2.8 |
| 例6 | 15岁 | HC+螺内酯 | 3个月 | 175.0 | 无 | 138/84 | B4P2 | 140/4.1 |
注:HC:氢化可的松;a同胞姐妹;bB:乳腺,P:阴毛;1 mmHg=0.133 kPa HC:Hydrocortisone;asisters;bB:breast,P:pubes
同期收治明确分型的新发CAH患儿共114例,其中21羟化酶缺乏症102例(89.5%),类脂性CAH 3例(2.6%),P450氧化还原酶缺乏症2例(1.7%),11β羟化酶缺乏症1例(0.9%)。6例17OHD占CAH总数的5.3%,位居第2位。
17OHD是CAH的罕见类型,约占CAH的1%[1],在某些特定族群中更常见,巴西和中国人发病率更高[2]。本中心17OHD例数占同期新诊断CAH例数的5.3%,是CAH除21羟化酶缺乏症外第二常见类型,既往认为排在第2位的11β羟化酶缺乏症在本组所占比例最低。
17OHD的致病基因CYP17A1编码产物包含两部分功能[3]:17α-羟化酶及17,20碳链裂解酶。绝大部分的CYP17A1基因突变可使酶功能联合受累,导致皮质醇和性激素合成不足,同时酶底物孕酮大量堆积,而17-OHP不高,ACTH、LH、FSH负反馈性升高。升高的ACTH刺激肾上腺增生,促使去氧皮质酮(DOC)及皮质酮大量增加,DOC在机体有强大的理盐和理糖作用,代偿皮质醇分泌不足的同时形成继发性高血压和低钾血症等[4]。本组6例患儿,临床可见高血压和/或低血钾,女性化外观,性腺功能低下;皮质醇水平低,ACTH轻中度高,孕酮升高,17-OHP及DHEA低下,CT示肾上腺增生,临床诊断17OHD成立。本病呈慢性高血压经过,儿童期高血压脏器损害不明显,提醒儿科医师应加强低血钾等可疑患儿的血压监测。对高血压女童常规行性激素六项检查,孕酮升高而17-OHP不高为本病的重要线索。
CYP17A1在人体的肾上腺及性腺表达[3],故17OHD不仅影响肾上腺功能,同时也影响性腺功能。46,XX者子宫卵巢发育不良,而46,XY者胚胎期的睾丸组织发育正常,可分泌抗苗勒氏管激素,故无子宫卵巢发育,但睾丸合成雄激素能力受损,导致其外生殖器发育障碍。绝大部分患者为女性外观,罕见男性尿道下裂及隐睾[5]。
目前中国发病患者绝大部分年龄为4~48岁[5,6,7,8],儿童时期就诊原因以高血压、低血钾为主。本研究中患儿年龄最小者仅1岁6个月,其就诊主因一侧睾丸位于阴唇下而致阴唇包块。与此发病类似,Çamtosun等[9]报道1例46,XY女童在2.5岁因女性外生殖器和腹股沟肿胀而入院,当时无高血压及低钾血症,被诊断为睾丸激素合成缺陷,随访至9岁时因血钾正常偏低而检查得以确诊。本组病例中,例6在5岁时也曾切除腹股沟包块后证实为睾丸组织,但未进一步检查导致诊断延误。提醒各级医师应加强对婴幼儿外生殖器检查,可能使46,XY的17OHD患儿尽早得到诊治。Hinz等[4]总结17OHD确诊年龄特点:88%为青春期,4%为婴幼儿期。本病确诊年龄较大的主要原因:(1)婴幼儿期肾小管对盐皮质激素不敏感,高血压及低血钾症状出现晚[3];(2)大量增加的DOC及皮质酮有糖皮质激素作用,肾上腺危象很少发生[10]。有关17OHD合并肾上腺危象的报道仅见于Zhang等[11]的报道:患儿临床表现为血压下降、低血糖及意识改变等。与经典21羟化酶缺乏症相比,17OHD患者的肾上腺增生程度不重,可能与大量堆积的DOC及皮质酮代偿皮质醇不足,导致ACTH轻度升高甚至正常水平有关。本组病例的肾上腺增生程度与临床症状严重程度无相关性。理论上由于DOC升高导致水钠潴留、高血压,进而抑制肾素及醛固酮的分泌,但本研究中肾素、醛固酮水平升高者更常见。多个病例系列报道均可见此现象,原因尚不清楚,Hinz等[4]总结了4种可能的高醛固酮血症假说:(1)17羟化酶活性部分缺乏导致肾素抑制不足;(2)皮质醇与醛固酮的负相关提示酶活性的严重缺乏;(3)皮质酮等前体的过度产生及转化;(4)增多的盐皮质激素前体干扰醛固酮检测。46,XY患者的睾丸位置较高,癌变风险高,故主张睾丸早期切除,但若17α-羟化酶/17,20-碳链裂解酶活性为部分丧失者,青春期有自发性性激素合成改善其青春期性发育,故切除睾丸时机可根据基因突变类型于青春期后抉择。
基因分析技术的发展及临床广泛应用,为17OHD的确诊提供了可能性和可行性。与17OHD相关的CYP17A1基因定位于10q24.3,包括8个外显子和7个内含子,编码508个氨基酸。目前已检出140多种突变,包括错义突变、剪接突变、插入缺失突变、同义突变及大的缺失等。p.Y329fs在本研究中出现最频繁,占等位基因的58.3%。与此一致,Zhang等[8]在26例中国人中检测到p.Y329fs占等位基因的53.8%,Han等[5]检测15例中国人p.Y329fs突变占46.7%。p.Y329fs是第6外显子第329位密码子的TAC→AA突变导致其后的移码突变,并提前产生终止密码子,生成一417个氨基酸的截短蛋白,丧失酶最重要的功能区。文献中该突变表述不同,易混淆。以下4种描述1045del,1047C>A;6436-6438TAC→AA;C.985-987delinAA;985缺失TAC,插入AA均为p.Y329fs。例1和例5为该基因纯合突变,例1患儿ACTH试验后皮质醇水平无升高,HCG试验后睾酮无升高,结合2例患儿的类固醇激素水平、性腺功能提示17α-羟化酶/17,20-碳链裂解酶活性完全丧失。外显子1-7缺失理论上可导致酶活性完全丧失。土耳其的1例完全型17OHD患者中可见外显子1-6纯合缺失报道[12]。8号外显子p.L465P变异在中国人群首次报道,既往也仅见Dhir等[13]报道1例15岁女童因原发性闭经就诊,乳房B4期,超声可见发育不良的子宫卵巢。进一步证实p.L465P导致17α-羟化酶活性剩余3%±1%。酶活性的部分缺失可解释青春期乳腺发育。8号外显子c.1246C>T(p.R416C)突变仅见于另一中国家系的2例同胞[14],已证实类似突变p.R416H可导致酶活性完全缺失[15],提示416位氨基酸是保证酶活性的重要位点。中国人另一热点突变D487-F489del未见于本组患儿。一项26例患者的临床及酶活性分析提示基因型与表型部分相关[8]。本病基因型和表型的关系尚不确切,酶活性完全丧失的患者,血压和血钾也可能正常。本研究中例2、4来自同一家庭,具有相同的基因突变,临床严重程度也不相同。似乎相同基因型患者46,XY核型者表现更为突出,越能早期得到诊治。
总之,17OHD的报道多见于成人,儿科医师对本病认识不足,多为单个病例报道。本研究分析总结6例患儿的临床及基因突变特点,有助于早期诊治此类患儿。
所有作者均声明不存在利益冲突


























