
探讨HSD17B4突变相关的过氧化物酶体D-双功能蛋白缺乏症(PDBPD)的临床与基因特征。
回顾性分析2020年8月南京医科大学附属儿童医院收治一家系2例HSD17B4突变致PDBPD的临床及基因情况。
男性先证者及同胞姐姐均伴新生儿癫痫、精神运动发育障碍、共济失调、肌无力、听力损害、足部畸形,血清极长链脂肪酸正常,头颅磁共振成像(MRI)示双侧小脑半球萎缩,肌电图:多发性周围神经神经源性损害肌电改变,听觉诱发电位:重度双侧感音神经性听力损失。基因检测显示HSD17B4复合杂合突变(c.1171G>C,c.686-2A>T),临床确诊为PDBPD,年龄分别为8岁和14岁。
报道2例HSD17B4突变致PDBPD,均符合典型临床表现。新发现HSD17B4基因c.1171G>C和c.686-2A>T突变,丰富了HSD17B4突变谱。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
过氧化物酶体D-双功能蛋白缺乏症(peroxisome D-bifunctional protein deficiency,PDBPD)又称为D-双功能蛋白缺乏症(D-bifunctional protein deficiency,DBPD),是一种罕见的常染色体隐性遗传病;发病率约为1:100 000[1]。国内尚未见相关报道。本研究对2020年8月南京医科大学附属儿童医院神经内科收治的一家系2例DBPD患儿的临床资料进行回顾性分析,并复习相关文献,以期提高对本病的认识。
先证者,男,8岁;先证者姐姐,女,14岁,均以"出生后精神运动发育迟缓"为主诉就诊。先证者系第2胎,第2产,足月剖宫产,出生体质量4.8 kg,身长55 cm;先证者姐姐系第1胎,第1产,足月顺产,出生体质量2.7 kg,身长46 cm。出生时均无窒息抢救史。先证者8月龄不会翻身,不会独坐,抓捏不佳;1.5岁会独走,走路不稳,行走姿势无明显异常;3岁后出现行走倒退,进展缓慢,目前能独走,步态不稳,姿势异常;康复治疗无明显进步,不会跑跳,上下楼梯需扶手。智力障碍,1岁左右会说话,口齿不清;2岁左右因"口齿不清"行舌系带手术,术后说话清晰度无改善;目前可与人简单交流,能说简单句子;不能接受适龄儿童教育,间断特殊幼儿园学习。其姐较先证者运动、认知、语言更差,自幼不会爬、走路,坐轮椅,抬头稳,独坐需靠东西,不能翻身,搀扶站立困难;目前仅会喊"爸爸、妈妈",口齿不清,可以理解部分简单语句,未上学。先证者及其姐均出生1 d出现抽搐,先证者表现为双手轻微节律性动,持续数十秒缓解,当地住院期间发作数次,予苯巴比妥治疗,未再抽搐发作,口服4年停用;其姐表现为口吐白沫,面色发绀,双眼上翻,四肢抖动,持续约3 min自行缓解,3~5次/d,发作2 d后当地医院住院,未行抗癫痫治疗,6月龄时再次发作1次,后未再发作。先证者出生时B超示肝大,卵圆孔未闭;6月龄及3~4岁头颅磁共振成像(MRI)未见异常;8月龄肌电图无异常。其姐有双侧髋关节脱位病史。其姐6岁肌电图:多发性周围神经源性损害肌电改变(运动、感觉纤维均受累)。父母体健,均37岁,非近亲婚配,否认类似病史。
生命体征正常,先证者体质量28 kg,身高130 cm;其姐体质量42 kg,身高145 cm。神志清,精神状态可,发育全面落后,口齿不清,双侧瞳孔等大等圆,直径3 mm,对光反射存在。斜视,双眼球活动可,眼球震颤,高腭,咽反射存在,颈软,气管居中,心肺腹未见异常;脊柱侧弯(其姐);四肢肌张力低下,肌力4级,双上肢手腕内旋(先证者),双手握力差,精细动作较同龄儿差,马蹄形内翻足(先证者);足趾活动障碍,不能完成足趾头背伸、屈曲、内收、外展动作(先证者);双足外翻(其姐),踝关节可被动过度屈伸(其姐);先证者共济运动检查阳性,其姐共济运动检查不能完成;独站不稳,步态不稳,姿势异常(先证者);其姐坐轮椅,扶站困难。先证者双侧膝反射活跃,其姐双侧膝反射减弱,先证者巴氏征阳性,其姐巴氏征阴性;踝阵挛阳性,跟腱挛缩(先证者)。感觉存在。
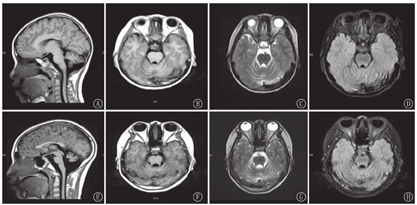
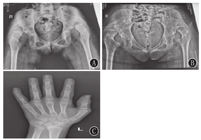
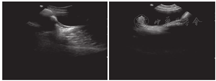
血尿便常规、血生化、甲状腺功能、脑脊液、胸片、心电图、肝胆胰脾+双肾上腺B超、染色体核型、尿有机酸分析及血氨基酸、肉碱和琥珀酰丙酮检验均无异常。雌二醇、孕酮、睾酮、催乳素、促卵泡生成素、促黄体生成激素均正常;血清极长链脂肪酸(VLCFA)正常。肌电图:多发性周围神经源性损害肌电改变(主要累及感觉、运动神经脱髓鞘)。听觉诱发电位:重度双侧感音神经性听力损失;先证者脑电图:异常儿童脑电图(背景活动减慢);其姐脑电图:正常青少年脑电图;先证者8岁头颅MRI:小脑脑沟宽深。其姐14岁头颅MRI:双侧小脑半球萎缩(图1)。先证者左腕关节正位片(骨龄片)正常。其姐14岁骨龄片:8枚腕骨发育,相当于10~11岁(图2)。先证者双肾B超正常;其姐双肾B超:双肾形态略小(左肾88 mm×39 mm,右肾85 mm×36 mm)。其姐14岁子宫附件B超:子宫形态小,宫体长经20 mm,前后径7 mm,横经10 mm,双侧卵巢显示不清(图3)。
经医院医学伦理委员会批准(批准文号:202011083-1)以及患儿监护人知情同意后,抽取先证者及其父母、其姐的外周血各2 mL行全外显子组测序,并对先证者及其父母、其姐进行Sanger检测及验证。结果示HSD17B4突变(c.686-2A>T,c.1171G>C),分别位于10、14外显子,经家系验证为复合杂合突变(图4、图5)。
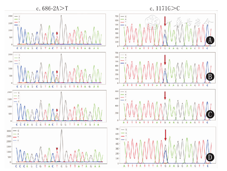
注:先证者及其姐HSD17B4复合杂合变异;左侧c.686-2A>T突变,父亲杂合突变,母亲正常;右侧c.1171G>C突变,父亲正常,母亲杂合突变 HSD17B4 compound heterozygous mutation was found in the proband and her sister;c.686-2A > T mutation in the left side,father heterozygous mutation and mother normal;c.1171G > C mutation in the right side,father normal and mother heterozygous mutation
以"HSD17B4"、"突变/变异"、"D-双功能蛋白缺乏/DBPD"、"测序"、"c.1171G>C/c.686-2A>T"及"HSD17B4""mutation"、"D-bifunctional protein deficiency/DBP deficiency"、"Sequencing"、"c.1171G>C/c.686-2A>T/c.686A>T"为关键词,查阅万方、中国知网、HGMD及PubMed数据库,时间自建库至2020年9月,共收集16篇文献报道HSD17B4突变相关病例,共163例HSD17B4突变致病患者,发现61种突变(缺失、插入、无义及错义突变)[1,2,3,4,5,6,7,8,9,10,11];美国、加拿大、澳大利亚、以色列、韩国、日本等DBPD病例相关的HSD17B4突变涉及c.650A>G和c.1704T>A、c.293A>AG、c.58+1G>A和c.680A>G、c.1132G>T和c.661C>T、c.1537C>A和c.1628G>C、c.101C>T和c.1547T>C、c.752G>A和c.868+1delG、c.14T>G和c.752G>A、c.1619A>G、c.46G>A、c.1369A>G和c.1516C>T、c.350A>T和c.836T>C、c.394C>T和c.523G>A等[3,4,5,6,12,13,14]。DBPD发病年龄差异较大,从新生儿期至中年均有报道;临床表型多样,包括98%(83/85例)新生儿低张力,93%(78/84例)新生儿低张力和癫痫发作,93%(79/85例)新生儿癫痫,34%(21/61例)视觉丧失,44%(27/61例)生长发育障碍,45%(29/64例)听力丧失,11%(7/61例)运动丧失,26%(18/69例)肝病,68%(53/79例)外部畸形,33%(4/12例)肾囊肿,42%(5/12例)肾上腺皮质萎缩。脑部影像学显示29%(14/47例)脑室系统粗大扩张,27%(13/47例)皮质发育不良,34%(16/47例)白质成熟延迟,17%(8/47例)大脑半球脱髓鞘,7%(3/47例)小脑半球脱髓鞘,17%(8/47例)小脑萎缩。脑尸检证实:45%(5/11例)脑发育不全/萎缩,64%(7/11例,外侧裂为主)多发性小脑回,55%(6/11例)胼胝体发育不良或萎缩,27%(3/11例)小脑发育不良,36%(4/11例)白质异位神经元,27%(3/11例)胶质增生,36%(4/11例)大脑脱髓鞘,27%(3/11例)小脑脱髓鞘。临床生理学显示:76%(96/126例)视网膜电图活动减弱,70%(88/126例)双侧脑干诱发电位减弱,70%(88/126例)神经传导速度减慢[1,3]。生化检测特征为血清VLCFA水平升高,少数(<2%)正常[1,4,9,11]。
DBPD是一种罕见的过氧化物酶体单个酶缺陷遗传病[2]。1989年由Watkins等[15]首次描述;1997年明确HSD17B4为其致病基因[16] 。本病以新生儿脑病、多发性神经病、精神运动发育障碍、双侧感音神经性听力损失、肝大、多发畸形为临床特征。目前还发现其他的临床表型,如卵巢功能障碍、小脑功能障碍、运动退化、骨成熟延迟、骨骼畸形等[1,3,4,6,7,8,9,11,17]。本组2例患儿为同胞姐弟,相同表型为新生儿癫痫发作、肌无力、精神运动发育迟缓、共济失调、足部畸形;姐姐还合并卵巢功能障碍、骨骼发育异常。临床电生理表现为双侧感音神经性听力损失、多发性周围神经病变。颅脑MRI示小脑萎缩,符合DBPD的诊断。患儿血清VLCFA正常,推测原因为酶活性残留。2例患儿严重程度有差异,推测原因为存在不同的酶活性残留量。
D-双功能蛋白是一种参与过氧化物酶体脂肪酸β氧化的酶,催化脂肪酸和脂肪酸衍生物的过氧化物酶体β氧化的第二(水合)和第三(脱氢)反应,由3个功能单元组成:(1)2-烯酰基辅酶A(CoA)水合酶单元;(2)3-羟酰基CoA脱氢酶单元;(3)甾醇载体蛋白-2-样单元[10]。根据所涉及的功能单元,DBPD分3种类型:2-烯酰基CoA水合酶和3-羟酰基CoA脱氢酶缺陷(Ⅰ型)、2-烯酰CoA水化酶缺陷(Ⅱ型)、3-羟酰基CoA脱氢酶缺陷(Ⅲ型);其中Ⅲ型最常见(45%),其次为Ⅱ型(28%)和Ⅰ型(27%)[1]。这3种类型表型异质性小,严重程度差异大;多数患儿(>80%)在2岁前死亡。Ⅰ型通常不超过2岁;Ⅱ和Ⅲ型倾向于存活超过10岁[1]。本组2例患儿未能行酶活性检查。研究显示,人类HSD17B4基因定位于染色体5q2,含24个外显子,3-羟酰基CoA脱氢酶单元由外显子1-12编码,2-烯酰基CoA水合酶单元由外显子12-21编码[1,3,10],本组2例基因突变位于10、14外显子,推测患儿属Ⅰ型可能性大。
迄今,国外学者报道了100多例HSD17B4突变所致的DBPD病例;鉴定了数十种HSD17B4突变[1,10]。国内尚未见相关报道。本研究通过全外显子组检测,发现HSD17B4复合杂合突变,突变位点为c.686-2A>T和c.1171G>C,分别来自父母双方,符合常染色体隐性遗传规律,美国医学遗传学和基因组学学会指南[18,19]评级为致病性变异。HSD17B4突变导致过氧化物酶体脂肪酸β氧化的破坏是其主要发病机制;β氧化底物VLCFA的累积是DBPD最重要的特征之一[1,2]。当酶活性残留,VLCFA正常时,脑白质和小脑白质脑病、脑皮质发育异常等"过氧化物酶体模式"可作为DBPD的诊断线索[20,21]。HSD17B4突变的病例回顾性研究显示,VLCFA正常时,部分病例可仅见小脑萎缩,以双侧感音神经性听力损失和女性独有的性腺发育不良为典型临床特征[7,11]。这些特征支持本组2例患儿诊断DBPD。
本病目前无特效治疗,以对症支持治疗为主,早期诊断、早期干预并进行相关的风险评估,可最大程度地降低其对患者影响。对于临床疑似DBPD患者,应注意进行详细的临床检查,评估生殖、骨骼、神经系统、听力情况等,重视神经电生理、颅脑MRI和相关生化检测,此外还要重视遗传分子生物学诊断,明确致病基因。
综上,本研究报道2例HSD17B4突变致DBPD,临床主要特征为新生儿癫痫发作、精神运动发育障碍、共济失调、肌无力、听力损害、多发畸形;女性可合并卵巢功能障碍。新发现的c.1171G>C和c.686-2A>T突变丰富了HSD17B4基因突变谱。本病尚无有效治疗与预防方法,早期明确诊断有助于遗传咨询和产前诊断。
所有作者均声明不存在利益冲突

























