
先天性肌无力综合征(CMS)是一组由遗传缺陷导致的神经肌肉信息传递障碍疾病,属于神经肌肉接头疾病。主要表现为骨骼肌疲劳性肌无力,大多数发病年龄早,常见死因为呼吸衰竭,致残率高。随着基因测序技术的提高及有关致病蛋白结构和功能的深入研究,过去20多年来对该病的发病机制有了更多认识,早期诊断和治疗可明显改善患者的症状。现针对CMS的病因、临床特点及诊疗等方面的研究进展进行综述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
先天性肌无力综合征(congenital myasthenic syndromes,CMS)是一组由遗传缺陷导致的神经肌肉传递障碍疾病,属于神经肌肉接头(NMJ)疾病[1,2]。20世纪70年代CMS作为一种特定的神经肌肉疾病开始被认识,其确切患病率不详,估计为重症肌无力的1/10,有可能更高;在英国每100万18岁以下儿童中,约有9.2例遗传确诊病例[3]。CMS主要表现为骨骼肌疲劳性肌无力,神经电生理检查重复神经电刺激(RNS)示波幅递减,单纤维肌电图(SFEMG)示异常颤抖或阻滞,血清抗乙酰胆碱受体(AChR)抗体和抗骨骼肌特异性受体酪氨酸激酶(MuSK)抗体等抗体阴性。由于遗传缺陷的分子机制不同,发病年龄、症状、肌无力受累部位及对治疗的反应各不相同。根据突变基因编码蛋白的表达位置,分为突触前综合征、突触基底层相关综合征、突触后综合征、糖基化缺陷等亚型。早期诊断和治疗可以明显改善患者的症状及预后,因此,本综述将针对CMS的病因、临床特点及诊疗等方面的研究进展进行系统阐述,以期患者得到早期诊断和治疗。
目前为止,已有30多个CMS致病基因被发现(表1)[3,4,5,6]。其中,编码AChR的基因(CHRNA1、CHRNB1、CHRND、CHRNE)突变最为常见,约占50%,RAPSN、DOK7、COLQ基因突变分别占10%~15%,CHAT基因突变约占5%[1,2]。随着测序技术的发展,未来有望发现更多的致病基因。基因突变导致的一种或多种形式的NMJ损害均可降低神经肌肉传递障碍的安全区间,即终板电位(EPP)与激活突触后电压门控Nav1.4通道触发肌纤维动作电位的阈值之间的差异。EPP振幅取决于各个突触囊泡中乙酰胆碱分子数量,神经冲动释放的突触囊泡数量,EPP量子含量,突触间隙中乙酰胆碱酯酶(AChE)活性,终板空间结构,AChR的密度、分布、动力学以及Nav1.4通道的密度和动力学等[1,2]。
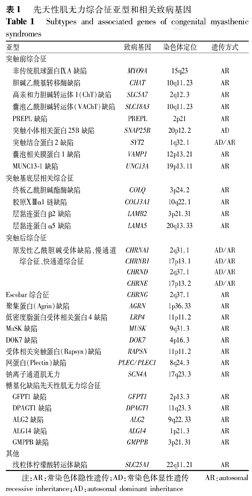
先天性肌无力综合征亚型和相关致病基因
Subtypes and associated genes of congenital myasthenic syndromes
先天性肌无力综合征亚型和相关致病基因
Subtypes and associated genes of congenital myasthenic syndromes
| 亚型 | 致病基因 | 染色体定位 | 遗传方式 | |
|---|---|---|---|---|
| 突触前综合征 | ||||
| 非传统肌球蛋白ⅨA缺陷 | MYO9A | 15q23 | AR | |
| 胆碱乙酰基转移酶缺陷 | CHAT | 10q11.23 | AR | |
| 高亲和力胆碱转运体1(ChT)缺陷 | SLC5A7 | 2q12.3 | AR | |
| 囊泡乙酰胆碱转运体(VAChT)缺陷 | SLC18A3 | 10q11.23 | AR | |
| PREPL缺陷 | PREPL | 2p21 | AR | |
| 突触小体相关蛋白25B缺陷 | SNAP25B | 20p12.2 | AD | |
| 突触结合蛋白2缺陷 | SYT2 | 1q32.1 | AD/AR | |
| 囊泡相关膜蛋白1缺陷 | VAMP1 | 12p13.21 | AR | |
| MUNC13-1缺陷 | UNC13A | 19p13.11 | AR | |
| 突触基底层相关综合征 | ||||
| 终板乙酰胆碱酯酶缺陷 | COLQ | 3p24.2 | AR | |
| 胶原ⅩⅢα1链缺陷 | COL13A1 | 10q22.1 | AR | |
| 层黏连蛋白β2缺陷 | LAMB2 | 3p21.31 | AR | |
| 层黏连蛋白α5缺陷 | LAMA5 | 20q13.33 | AR | |
| 突触后综合征 | ||||
| 原发性乙酰胆碱受体缺陷、慢通道综合征、快通道综合征 | CHRNA1 | 2q31.1 | AD/AR | |
| CHRNB1 | 17p13.1 | AD/AR | ||
| CHRND | 2q37.1 | AD/AR | ||
| CHRNE | 17p13.2 | AD/AR | ||
| Escobar综合征 | CHRNG | 2q37.1 | AR | |
| 聚集蛋白(Agrin)缺陷 | AGRN | 1p36.33 | AR | |
| 低密度脂蛋白受体相关蛋白4缺陷 | LRP4 | 11p11.2 | AR | |
| MuSK缺陷 | MUSK | 9q31.3 | AR | |
| DOK7缺陷 | DOK7 | 4p16.3 | AR | |
| 受体相关突触蛋白(Rapsyn)缺陷 | RAPSN | 11p11.2 | AR | |
| 网蛋白(Plectin)缺陷 | PLEC/PLEC1 | 8q24.3 | AR | |
| 钠离子通道肌无力 | SCN4A | 17q23.3 | AR | |
| 糖基化缺陷先天性肌无力综合征 | ||||
| GFPT1缺陷 | GFPT1 | 2p13.3 | AR | |
| DPAGT1缺陷 | DPAGT1 | 11q23.3 | AR | |
| ALG2缺陷 | ALG2 | 9q22.33 | AR | |
| ALG14缺陷 | ALG14 | 1p21.3 | AR | |
| GMPPB缺陷 | GMPPB | 3p21.31 | AR | |
| 其他 | ||||
| 线粒体柠檬酸转运体缺陷 | SLC25A1 | 22q11.21 | AR | |
注:AR:常染色体隐性遗传;AD:常染色体显性遗传 AR:autosomal recessive inheritance;AD:autosomal dominant inheritance
CMS的特征表现是骨骼肌(如眼肌、球肌、四肢肌肉)疲劳性无力,认知大多正常。大多在出生时、生后不久或儿童早期发病,也有部分直到儿童后期才出现症状,甚至在成年后出现。心脏和平滑肌通常不受累。疾病的严重程度和病程不一,从症状轻微到进行性致残性无力。在CMS的某些亚型中,肌无力症状可能轻微,但发热、感染、兴奋或过度运动可能会导致肌无力突然严重、恶化,甚至呼吸衰竭突然发作。
肌酸激酶正常或轻度升高;抗AChR抗体、抗MuSK抗体、抗AChE抗体等抗体均阴性,这是诊断CMS的必要条件。
RNS示波幅递减10%以上。一般情况下,低频(2~3 Hz)RNS即可出现波幅递减,部分未受累的肌肉在静息状态下也可不出现波幅递减改变,但以10 Hz频率长时间(5~10 min)刺激骨骼肌仍可诱发出显著波幅递减现象。SFEMG示异常颤抖或阻滞。运动神经传导检测中单个神经刺激出现重复复合肌肉动作电位(CMAP)是慢通道综合征、终板AChE缺陷CMS的特征性表现,也可见于服用大剂量胆碱酯酶抑制剂患者。
可通过静脉注射依酚氯铵或肌内注射甲基硫酸新斯的明或口服胆碱酯酶抑制剂对照观察试验进行评估。对儿童来说,一般选择甲基硫酸新斯的明试验。对于仅有疲劳性肌无力而无眼睑下垂、球肌受累表现的患者,也可通过口服胆碱酯酶抑制剂对照观察试验进行评估。
依据临床特征及辅助检查,可做出临床诊断,确诊需要进行基因检测。当临床研究指向某个/某些候选基因时(表2),对特定CMS的遗传诊断有很大帮助,可选择单基因测序或靶向捕获二代测序[1,2]。

先天性肌无力综合征基因诊断线索
Genetic diagnosis clues of congenital myasthenic syndromes
先天性肌无力综合征基因诊断线索
Genetic diagnosis clues of congenital myasthenic syndromes
| 临床表现 | 候选致病基因 |
|---|---|
| 由感染、发热等诱发的发作性呼吸暂停 | CHAT、RAPSN、SLC5A7、SLC18A3、SCN4A |
| 肢带型肌无力 | DOK7、MUSK、GFPT1、DPAGT1、ALG2、ALG14、GMPPB,偶见RAPSN和COLQ |
| 认知障碍 | DPAGT1、SNAP25B |
| 先天性多发性关节挛缩 | RAPSN、CHRNE或CHRNG、SNAP25B |
| 新生儿或婴儿期喘鸣、声带麻痹 | DOK7 |
| 单纯大疱性表皮松解症、肌营养不良 | PLEC |
| 肾病综合征、眼畸形 | LAMB2 |
| 瞳孔对光反射迟钝 | 部分COLQ |
| 单个神经刺激出现重复CMAP | COLQ和CHRNA1、CHRNB1、CHRND、CHRNE导致的慢通道先天性肌无力综合征 |
| 持续5 min 10 Hz RNS,CMAP和EPP振幅递减50%以上,并在至少5 min后缓慢逐渐恢复 | CHAT |
| 肌肉病理管聚集现象 | GFPT1、DPAGT1、ALG2 |
| 自噬性肌病 | GFPT1、DPAGT1 |
| 常染色体显性遗传 | CHRNA1、CHRNB1、CHRND、CHRNE导致的慢通道先天性肌无力综合征,SNAP25B,部分SYT2 |
| 胆碱酯酶抑制剂无反应或加重 | CHRNA1、CHRNB1、CHRND、CHRNE导致的慢通道CMS,COLQ,LAMB2,DOK7,MUSK,AGRN,LRP4,PLEC |
注:CMAP:复合肌肉动作电位;RNS:重复神经电刺激;EPP:终板电位 CMAP:compound muscle action potential;RNS:repetitive electrical nerve stimulation;EPP:end-plate potential
CMS需与重症肌无力、肉毒中毒、脑干病变、先天性肌病、先天性肌营养不良、肢带型或面肩肱型肌营养不良、婴儿强直性肌营养不良、脊肌萎缩症、线粒体肌病、先天性眼外肌纤维化、Möbius综合征等进行鉴别[1,2]。
根据发病机制,可将突触前综合征分为影响轴突运输(MYO9A)、乙酰胆碱合成和回收(CHAT、SLC5A7、SLC18A3、PREPL)、突触囊泡胞吐(SNAP25B、SYT2、VAMP1、UNC13A),目前共有9个致病基因。这组患者临床表现相似,多表现为发病早且病情重,发作性呼吸暂停,以及由于编码蛋白表达在中枢神经系统而引起的中枢神经系统症状[3]。溴吡斯的明和/或3,4-二氨基吡啶(3,4-DAP)治疗有效。CHAT突变导致的胆碱乙酰基转移酶(ChAT)缺陷是最常见的突触前综合征。
ChAT由CHAT基因编码,催化乙酰辅酶A和胆碱到乙酰胆碱的合成。2001年CHAT基因突变被首次报道导致CMS伴发作性呼吸暂停(CMS-EA),通常表现为新生儿期或婴儿期发病,眼肌、球肌、肢体肌肉无力、易疲劳,肌张力低下、呼吸衰竭伴发作性呼吸暂停和发绀、喂养困难等,症状可能由感染、发热、兴奋或过度运动等诱发或加重[7,8]。呼吸暂停发作常被误认为抽搐发作,需注意鉴别,另外,其可引起突然死亡或缺氧性脑损伤,需引起临床医师高度警惕。发作性呼吸暂停是CMS-EA的一个特征性表现,后来的研究发现,其也可出现于胆碱转运体1(ChT)缺陷[9]、囊泡乙酰胆碱转运体(VAChT)缺陷[10]、钠通道型肌无力[11]和Rapsyn缺陷[12,13]等亚型中。CHAT突变的致病机制包括改变了ChAT的表达水平、催化效率和结构稳定性[14]。最严重的动力学异常通常由酶活性位点通道中的突变、靠近底物结合位点的突变、或发挥其变构作用的突变所引起[1,14],大片段缺失突变患者的表型可能更重[15]。
神经电生理检查对CHAT基因突变的诊断具有重要提示意义,虽然低频(2~3 Hz) RNS在某些患者无递减,但持续5 min高频(10~20 Hz) RNS CMAP和EPP振幅可递减50%以上(正常递减<30%),并要在停止刺激后至少5 min才逐渐恢复[1]。此项检查在儿科需在麻醉状态下进行,对高度疑似患者尽早行基因检测有助于早期诊断。
胆碱酯酶抑制剂对大部分患者有效,少数患者存在顽固的呼吸暂停反复发作,需持续性呼吸支持[7]。指导父母使用便携式呼吸器、家用呼吸暂停监护仪和肌内注射甲基硫酸新斯的明在CMS-EA的管理中非常重要[2]。本病预后不良,一项较大样本长期随访研究表明,即便经过了长期规范的胆碱酯酶抑制剂治疗,患者最终需依赖轮椅生活[7]。
基底层是位于突触间隙的细胞外基质的一种结构形式,对突触前和突触后结构的排列、组织和维持至关重要。编码基底层蛋白的基因突变导致基底层结构发生改变,影响NMJ稳定性而致病。目前已发现4个致病基因,分别为编码AChE胶原锚定单位—胶原Q的COLQ,编码胶原ⅩⅢα1链的COL13A1,编码层黏连蛋白β2和α5的LAMB2和LAMA5。COL13A1、LAMB2、LAMA5突变较为罕见,下面以COLQ突变导致的终板AChE缺陷为代表进行详述。
COLQ编码终板AChE胶原锚定单位—胶原Q(ColQ),负责将AChE锚定在突触基底层,从而发挥水解乙酰胆碱的生理作用[16]。ColQ由3个结构域所组成:N-末端结构域的突变阻止胶原结构域与催化亚基结合;胶原结构域的突变截断并阻止三螺旋胶原结构域的组装;C-末端结构域的大多数突变会降低ColQ表达或阻止胶原结构域的三螺旋组装[1]。在这3个结构域中,突变均有报道。COLQ相关CMS可能通过不同机制的神经肌肉传递受损而致。COLQ突变引起AChE水解活动下降,乙酰胆碱在突触间隙存在时间延长,可导致AChR脱敏和由肌浆Ca2+超负荷所致的继发性终板肌病伴随AChR丢失。如果COLQ突变改变了ColQ与MuSK的相互作用,则可能影响突触后分化[17,18]。典型表现为新生儿期或婴幼儿期发病,临床表现重,眼睑下垂,眼肌麻痹,全身无力和呼吸困难,部分患者瞳孔对光反射迟钝[2]。C-末端结构域的错义突变相关病例可能儿童后期发病,临床表现较轻[19]。除了一般NMJ病变的神经电生理表现外,终板AChE缺陷CMS还具有单个神经刺激出现重复CMAP的特征性表现。这是由于乙酰胆碱在突触间隙存在时间延长,使EPP的持续时间延长,当EPP超过肌纤维的绝对不应期时,它会产生第2个肌纤维动作电位,即重复CMAP[2]。
治疗上,禁用溴吡斯的明和3,4-DAP,β2受体激动剂(麻黄碱和沙丁胺醇)治疗有效,但发挥疗效较慢,机制尚不清楚[1,19]。
肌肉烟碱型AChR有2个亚型:成人型(ε-nAChR)和胎儿型(γ-nAChR),成人型分布在终板区,由2 α、β、δ、和ε 5个亚基组成,分别由CHRNA1、CHRNB1、CHRND和CHRNE基因编码;胎儿型分布在终板区和终板区外,由2 α、β、δ、和γ 5个亚基组成,分别由CHRNA1、CHRNB1、CHRND和CHRNG基因编码[2]。在胚胎发育过程中,γ-nAChR参与了早期突触后膜结构的形成,在发育约30周后,γ亚基被ε亚基所取代[20]。原发性AChR缺陷的特征是AChR数量减少(正常值的10%~30%)和突触后皱褶完整性破坏[3]。微电极研究显示微终板电位幅度降低[1]。原发性AChR缺陷CMS临床轻重不一,绝大部分患者存在眼睑下垂和眼球运动受限,伴中-重度肢体无力,眼球运动受限可作为基因诊断的一项重要线索[1,3]。任何一个AChR亚基的隐性错义、无义或剪接位点和启动子区突变均可导致原发性AChR缺陷,但大多数在ε亚基中检测到[1]。ε亚基突变频率高的原因为胎儿γ亚基可代替缺陷的ε亚基进行表型拯救,除了严重的眼睑下垂和眼肌麻痹外,大多数ε亚基低表达突变患者病程相对良性,而其他亚基由于不存在替代亚基,不能被拯救,携带CHRNA1、CHRNB或CHRND基因纯合或复合杂合无效突变的个体可能在宫内死亡,其他非ε亚基低表达突变患者在婴幼儿期发病,病情重,死亡率高[1,2]。治疗上,胆碱酯酶抑制剂和3,4-DAP有效,部分患者添加β2受体激动剂效果显著[1,3]。
AChR 5个亚基中的任何一个亚基缺陷均可能导致离子通道功能改变,引起离子通道开放后关闭减慢(慢通道综合征,SCCMS)或关闭加快(快通道综合征,FCCMS)。SCCMS潜在的致病机制为AChR开放时间延长,导致突触后膜持续去极化,形成去极化块,并致AChR脱敏,以及由于Ca2+超负荷引起的继发性终板肌病[1]。SCCMS为常染色体显性遗传,发病年龄不等(出生到50岁),症状一般没有其他亚型严重,但新生儿发病的一般较重,常累及颈部、肩胛和上肢远端肌肉,也可表现为眼睑下垂和眼肌麻痹,但程度低于原发性乙酰胆碱受体缺陷[3]。与终板乙酰胆碱酯酶缺陷CMS相似,单个神经刺激出现2个CMAP峰。不过,检测不到重复CMAP并不能绝对排除SCCMS的可能[21]。治疗上,禁用溴吡斯的明和3,4-DAP,可用AChR开放通道阻滞剂氟西汀或奎尼丁治疗[2,3]。
FCCMS与SCCMS相反,AChR通道开放概率下降,开放时间缩短,导致突触后去极化减少,不能触发肌肉动作电位的机会增加。FCCMS为常染色体隐性遗传,临床表现轻重不一,大多表现为生后严重的肌无力、眼睑下垂、眼肌麻痹、呼吸衰竭[22]。FCCMS罕见,AChR亚基缺陷为纯合或复合杂合突变,其中一个等位基因须为无效突变或低表达突变[3],这些突变位于AChR的各个结构域,导致对乙酰胆碱亲和力降低,门控效率降低,或通道动力学不稳定,或通过这些机制联合作用[2]。治疗上,溴吡斯的明和3,4-DAP有效,但随着时间的延长,治疗效果有可能减弱[3]。
AChR聚集信号传导通路在NMJ的形成和维持中必不可少,由一系列蛋白通过单向或多向的结合、激活、聚集等复杂的生理过程实现。目前比较清楚的经典通路是在神经末梢释放聚集蛋白(Agrin)到突触间隙后,与突触后膜上的低密度脂蛋白受体相关蛋白4(LRP4)结合,Agrin-LRP4复合体结合并活化MuSK,导致下游酪氨酸激酶7(DOK7)磷酸化,又反过来促进DOK7的全面激活,并募集其他衔接蛋白诱发受体相关突触蛋白(Rapsyn),使AChR在突触后膜聚集,增加突触特异性基因表达,促进突触后分化[23,24,25]。编码Agrin、LRP4、MuSK、DOK7、Rapsyn等蛋白的基因突变均可导致CMS[2],除RAPSN基因突变外,其他突变临床表现相似,主要为肢带型肌无力,溴吡斯的明和3,4-DAP可能无效,β2受体激动剂有效[3],其中,DOK7-CMS亚型最常见。
DOK7是MuSK的一种肌肉特异性胞质衔接蛋白,对神经肌肉突触形成至关重要[26]。DOK7缺陷为常染色隐性遗传,基因型广泛,但c.1124_1127dupTGCC移码变异最常见,至少在60%以上的病例中出现[27,28]。DOK7缺陷临床轻重不一,虽然没有明确的基因型-表型相关性,但患者均表现为肢带型肌无力,面部和颈部肌肉较少受累,部分患者有严重的球麻痹和眼肌不全性麻痹[1]。有些患者最初被误诊为肢带型肌营养不良,肌肉病理1型纤维占优势、2型纤维萎缩、轻度肌病性改变是其相关特征[1]。治疗上,禁用溴吡斯的明,β2受体激动剂有明显疗效[2,29]。
RAPSN编码43 kDa受体相关突触支架蛋白,将AChR聚集并锚定在突触后膜并对突触后膜皱褶的发育至关重要。Rapsyn通过其不同区域与其他分子的结合发挥其功能:N-末端结构域将Rapsyn连接到突触后膜,7个四肽重复序列与MuSK胞质区域结合,卷曲螺旋结构域与AChR亚基的细胞内环结合,C末端结构域与β抗肌萎缩蛋白聚糖的结合使Rapsyn与肌动蛋白细胞骨架相连[1]。RAPSN相关CMS的特点是突触后膜AChR缺乏和突触连接皱褶发育不良[12]。大多数患者表现为出生后或婴幼儿期全身性肌张力低下、呼吸困难和喂养困难;轻度关节挛缩、面部畸形,眼睑下垂和斜视也较为常见,但眼肌麻痹相对少见,危及生命的呼吸困难常发生在婴儿期和儿童早期,多由感染、发热诱发,导致缺氧性脑病。儿童晚期发病的患者临床病程通常较轻[13,30]。RAPSN相关CMS为常染色隐性遗传。p.N88K变异最为常见,特别在印度和欧洲人中[31],其他变异出现在启动子区和整个开放阅读框中。除了携带纯合E-box突变(c.-38A>G)的中东犹太患者表现为眼睑下垂、下颚突出、咀嚼肌和面部肌肉无力及高鼻音等较轻临床表型外[32],没有明确的基因型和表型的相关性。多数患者对胆碱酯酶抑制剂反应良好,部分患者添加3,4-DAP有效,部分患者在胆碱酯酶抑制剂和3,4-DAP效果欠佳时,可通过添加麻黄碱或沙丁胺醇获益。多数患者随着年龄的增长临床改善,长期预后良好[33]。
蛋白糖基化对新生蛋白的折叠、稳定、溶解、聚集和运输十分重要。目前已发现在5个编码糖基化酶的基因—GFPT1、DPAGT1、ALG2、ALG14和GMPPB中发现的突变而引起的在内质网早期蛋白N-连接糖基化缺陷与CMS有关[34,35,36,37,38]。关于NMJ功能受损的机制,起初认为在于糖基化酶催化功能下降或缺失导致AChR亚基糖基化异常,阻碍了AChR亚基向细胞表面的有效运输及其五聚体在突触后膜的正常组装,最终肌肉终板的AChR数目减少[3],但在确定了更多与糖基化缺陷有关的CMS患者后发现,糖基化缺陷可从突触前和突触后多方面损伤神经肌肉间的信号传递[1,35]。糖基化缺陷CMS通常表现为肢带型肌无力,随着病程的进展,常伴随肌病,而典型的受累部位眼肌、面肌通常不受累[3,34]。肌肉病理在多数患者中可以发现肌纤维内管聚集,是这组表型的一个诊断线索,但并非所有患者都有这种现象。溴吡斯的明和3,4-DAP治疗有效,部分可联合应用沙丁胺醇[2,3]。
CMS的治疗药物主要有胆碱酯酶抑制剂、钾离子通道阻滞剂、AChR阻滞剂和β2受体激动剂,免疫抑制剂治疗无效[39]。溴吡斯的明和3,4-DAP在数小时内发挥作用,而奎尼丁、氟西汀、麻黄碱和沙丁胺醇作用较慢,可能在数天、数周或数月内发挥作用[2]。
溴吡斯的明,通过抑制AChE分解乙酰胆碱而发挥作用。溴吡斯的明是CMS的一线治疗药物,除了CHRNA1、CHRNB1、CHRND、CHRNE突变导致的慢通道CMS,以及COLQ、LAMB2、DOK7、MUSK、AGRN、LRP4、PLEC基因突变有关的CMS外,其他亚型的CMS均首选溴吡斯的明治疗,其中,慢通道CMS、COLQ、DOK7、LRP4、LAMB2亚型禁用溴吡斯的明。
3,4-DAP阻断突触前膜快电压门控钾离子通道,延长突触前膜去极化,从而增加动作电位持续时间和乙酰胆碱释放。3,4-DAP是CMS的二线治疗药物,多作为溴吡斯的明的添加治疗,也在一些对胆碱酯酶抑制剂无反应如MUSK、PLEC突变中,可能有效[2]。
氟西汀、奎尼丁在慢通道综合征中作为长效通道阻滞剂缩短AChR通道开放时间,阻止突触后膜去极化块的形成和受体在生理条件下的脱敏。由于奎尼丁更容易引起心脏传导障碍,干扰依赖细胞色素P450ⅡDA通路的药物,在实践中氟西汀更为常用。然而,由于氟西汀有产生自杀意念不良反应的危险,儿童和青少年,必须在精神科监测下使用,大剂量可延长QT间期。
麻黄碱和沙丁胺醇对CMS的治疗作用机制不明。推测它们可稳定NMJ并降低AChR的分散。β2受体激动剂是COLQ、DOK7、MUSK、AGRN、LRP4突变的一线治疗药物,也在一些胆碱酯酶抑制剂治疗有效但剂量需求大的患者中作为添加治疗发挥作用,如原发性AChR缺陷、Rapsyn缺陷、糖基化缺陷CMS。
所有作者均声明不存在利益冲突

























