
探讨慢性肉芽肿病(CGD)患儿的治疗和预后情况,以加强临床医师对该病的管理,提高患儿生存质量。
纳入重庆医科大学附属儿童医院明确诊断为CGD且于1999年1月至2016年12月出生的82例患儿,分为存活组和死亡组,对其一般特征、感染、炎症并发症、治疗及生存情况进行分析。
82例患儿中存活组55例(67.1%),死亡组27例(32.9%)。49例(59.8%)新生儿期即出现症状,死亡组所占比例(81.5%)高于存活组(49.1%),差异有统计学意义(P<0.05)。确诊后58例(70.7%)开始服用复方磺胺甲 唑预防感染,平均用药36.8个月;52例(63.4%)接受伏立康唑或伊曲康唑预防真菌感染,平均用药34.3个月。82例患儿累计因感染住院288次,细菌、真菌、结核分枝杆菌三大病原检出率为41.3%,死亡组(51.8%)高于存活组(36.9%),差异有统计学意义(P<0.05)。检出细菌以肺炎克雷伯菌、金黄色葡萄球菌、洋葱伯克霍尔德菌、大肠埃希菌为主,其中肺炎克雷伯菌、大肠埃希菌均为超广谱β-内酰胺酶阳性菌。死亡组患儿使用特殊级抗生素比例(70.4%)高于存活组(40.0%),差异有统计学意义(P<0.05)。17例(20.7%)患儿出现炎症并发症,以间质性肺病或肺纤维化、炎症性肠病、不完全川崎病为主。32例(39.0%)患儿接受造血干细胞移植(HSCT),其中25例(78.1%)于重庆医科大学附属儿童医院移植,成功率为96.0%(24/25例)。Kaplan-Meier生存分析曲线显示新生儿期即出现症状、依赖于特殊级抗生素治疗、未接受移植的患儿生存率更低。
唑预防感染,平均用药36.8个月;52例(63.4%)接受伏立康唑或伊曲康唑预防真菌感染,平均用药34.3个月。82例患儿累计因感染住院288次,细菌、真菌、结核分枝杆菌三大病原检出率为41.3%,死亡组(51.8%)高于存活组(36.9%),差异有统计学意义(P<0.05)。检出细菌以肺炎克雷伯菌、金黄色葡萄球菌、洋葱伯克霍尔德菌、大肠埃希菌为主,其中肺炎克雷伯菌、大肠埃希菌均为超广谱β-内酰胺酶阳性菌。死亡组患儿使用特殊级抗生素比例(70.4%)高于存活组(40.0%),差异有统计学意义(P<0.05)。17例(20.7%)患儿出现炎症并发症,以间质性肺病或肺纤维化、炎症性肠病、不完全川崎病为主。32例(39.0%)患儿接受造血干细胞移植(HSCT),其中25例(78.1%)于重庆医科大学附属儿童医院移植,成功率为96.0%(24/25例)。Kaplan-Meier生存分析曲线显示新生儿期即出现症状、依赖于特殊级抗生素治疗、未接受移植的患儿生存率更低。
CGD患儿临床表现出现越早、特殊级抗生素使用比例越高,提示病情越重,死亡率越高;目前药物保守治疗仍有不足,耐药菌的产生更增加了治疗难度。炎症并发症的认识和治疗仍有待加强。HSCT是目前CGD患儿的唯一治愈方法。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
慢性肉芽肿病(chronic granulomatous disease,CGD)是一种罕见的原发性免疫缺陷病,由基因突变致吞噬细胞还原型辅酶Ⅱ(NADPH)氧化酶功能障碍引起。CGD主要表现为反复的细菌、真菌感染,异常的炎症反应和肉芽肿形成。目前与CGD相关的基因包括CYBB、CYBA、NCF1、NCF2、NCF4、CYBC1[1]。我国CGD发病率尚不清楚,国外报道的发病率约为1/250 000[2] ,大多数患者在5岁前确诊[3,4]。常见的治疗方法包括抗感染、γ-干扰素、造血干细胞移植(hematopoietic stem cell transplantation,HSCT)等,显著改善了CGD患者的预后。随着抗感染治疗的改进,炎症并发症的发生成为引起关注的新问题[5] 。我国在CGD患儿的管理方面仍面临挑战。本研究回顾性分析82例CGD患儿的感染、炎症并发症及治疗、预后等情况,为临床治疗和管理CGD患儿、提高患儿生存率提供一定依据。
研究纳入重庆医科大学附属儿童医院明确诊断且于1999年1月至2016年12月出生的82例CGD患儿。排除标准:(1)2016年12月后出生;(2)失访;(3)资料不全(包括3例脐血干细胞移植患儿)。本研究通过重庆医科大学附属儿童医院医学伦理委员会批准[批准文号:(2020)年伦审(研)第77号]。本研究为回顾性研究,不涉及患儿诊治干预,重庆医科大学附属儿童医院医学伦理委员会已通过免除签署知情同意书。
通过电子病历系统收集CGD患儿的病史资料,包括患儿的一般资料、感染病原及药敏结果、炎症并发症、治疗情况、生存情况等。一般资料包括出生日期、性别、诊断日期、生存状况、是否接种卡介苗及是否有接种反应、新生儿期是否有症状等。治疗包括抗感染(细菌、真菌、结核)治疗及HSCT的供体类型、预处理方案、移植物抗宿主病(GVHD)发生等情况。死亡分析主要包括死亡原因和病原学检测。
采用SPSS 22.0软件进行统计分析。连续变量以中位数、平均值、最小/最大值表示,连续变量比较用Mann-Whitney U检验。计数资料用例(%)表示。分类变量使用Pearson χ2检验或Fisher′s确切概率法进行比较。生存分析采用Kaplan-Meier非参数模型。P<0.05为差异有统计学意义。
82例CGD患儿中,男80例(97.6%),女2例(2.4%)。其中X连锁隐性遗传73例(89.0%)(CYBB基因突变,包含1例XK、CYBB、DYNLT3复合突变),常染色体隐性遗传6例(3例NCF1突变,3例CYBA突变),另有3例未发现突变基因。
存活组55例(67.1%),平均年龄70.0(24.9~232.0)个月;死亡组27例(32.9%),平均年龄37.6(2.0~190.3)个月。患儿平均起病年龄为3.7(0.1~24.0)个月,平均确诊年龄为26.3(0.6~165.3)个月。截止随访日期为2018年12月31日,中位随访时间25.9(0.1~164.0)个月。死亡组患儿的中位随访时间明显短于存活组(P<0.05),确诊后生存时间不足6个月。49例(59.8%)新生儿期即出现症状,死亡组所占比例显著高于存活组,差异有统计学意义(P<0.05)。70例(85.4%)接种卡介苗,有无卡介苗接种局部不良反应、卡介苗病(包括播散性卡介苗病)2组差异无统计学意义(表1)。
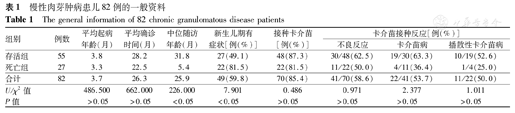
慢性肉芽肿病患儿82例的一般资料
The general information of 82 chronic granulomatous disease patients
慢性肉芽肿病患儿82例的一般资料
The general information of 82 chronic granulomatous disease patients
| 组别 | 例数 | 平均起病年龄(月) | 平均确诊时间(月) | 中位随访年龄(月) | 新生儿期有症状[例(%)] | 接种卡介苗[例(%)] | 卡介苗接种反应[例(%)] | ||
|---|---|---|---|---|---|---|---|---|---|
| 不良反应 | 卡介苗病 | 播散性卡介苗病 | |||||||
| 存活组 | 55 | 3.8 | 28.2 | 31.8 | 27(49.1) | 48(87.3) | 30/48(62.5) | 19/30(63.3) | 10/19(52.6) |
| 死亡组 | 27 | 3.3 | 22.5 | 5.4 | 22(81.5) | 22(81.5) | 11/22(50.0) | 4/11(36.4) | 1/4(25.0) |
| 合计 | 82 | 3.7 | 26.3 | 25.9 | 49(59.8) | 70(85.4) | 41/70(58.6) | 22/41(53.7) | 11/22(50.0) |
| U/χ2值 | 486.500 | 662.000 | 226.000 | 7.901 | 0.486 | 0.971 | 2.377 | 1.011 | |
| P值 | >0.05 | >0.05 | <0.05 | <0.05 | >0.05 | >0.05 | >0.05 | >0.05 | |
纳入CGD患儿的病原学检出情况见表2。分枝杆菌感染和真菌感染由于培养阳性率低,故根据临床表现、病原学(涂片、培养、PCR、G/GM试验等)及影像学综合诊断。以痰液标本中肺炎支原体(MP)-PCR≥5×105 copies/L或者血液中MP抗体≥1∶160提示MP感染。病毒感染主要为EB病毒(EBV)、巨细胞病毒、腺病毒感染。
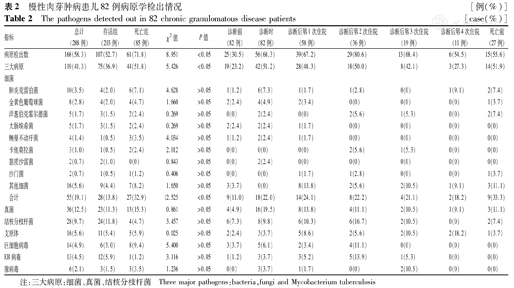
慢性肉芽肿病患儿82例病原学检出情况[例(%)]
The pathogens detected out in 82 chronic granulomatous disease patients [case(%)]
慢性肉芽肿病患儿82例病原学检出情况[例(%)]
The pathogens detected out in 82 chronic granulomatous disease patients [case(%)]
| 指标 | 总计(288例) | 存活组(203例) | 死亡组(85例) | χ2值 | P值 | 诊断前(82例) | 诊断时(82例) | 诊断后第1次住院(58例) | 诊断后第2次住院(36例) | 诊断后第3次住院(19例) | 诊断后第4次住院(11例) | 死亡前(27例) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 病原检出数 | 168(58.3) | 107(52.7) | 61(71.8) | 8.951 | <0.05 | 25(30.5) | 56(68.3) | 39(67.2) | 29(80.6) | 13(68.4) | 6(54.5) | 15(55.6) | |
| 三大病原 | 119(41.3) | 75(36.9) | 44(51.8) | 5.426 | <0.05 | 19(23.2) | 42(51.2) | 28(48.3) | 18(50.0) | 8(42.1) | 3(27.3) | 14(51.9) | |
| 细菌 | |||||||||||||
| 肺炎克雷伯菌 | 10(3.5) | 4(2.0) | 6(7.1) | 4.628 | >0.05 | 1(1.2) | 6(7.3) | 1(1.7) | 1(2.8) | 0(0) | 1(9.1) | 2(7.4) | |
| 金黄色葡萄球菌 | 8(2.8) | 4(2.0) | 4(4.7) | 1.660 | >0.05 | 2(2.4) | 4(4.9) | 2(3.4) | 0(0) | 0(0) | 0(0) | 1(3.7) | |
| 洋葱伯克霍尔德菌 | 5(1.7) | 3(1.5) | 2(2.4) | 0.269 | >0.05 | 0(0) | 2(2.4) | 0(0) | 2(5.6) | 1(5.3) | 0(0) | 2(7.4) | |
| 大肠埃希菌 | 5(1.7) | 3(1.5) | 2(2.4) | 0.269 | >0.05 | 2(2.4) | 2(2.4) | 1(1.7) | 0(0) | 0(0) | 0(0) | 0(0) | |
| 鲍曼不动杆菌 | 4(1.4) | 1(0.5) | 3(3.5) | 4.034 | >0.05 | 1(1.2) | 2(2.4) | 1(1.7) | 0(0) | 0(0) | 0(0) | 0(0) | |
| 卡他莫拉菌 | 3(1.0) | 1(0.5) | 2(2.4) | 2.012 | >0.05 | 0(0) | 0(0) | 0(0) | 2(5.6) | 1(5.3) | 0(0) | 0(0) | |
| 黏质沙雷菌 | 2(0.7) | 2(1.0) | 0(0) | 0.843 | >0.05 | 0(0) | 2(2.4) | 0(0) | 0(0) | 0(0) | 0(0) | 0(0) | |
| 沙门菌 | 2(0.7) | 1(0.5) | 1(1.2) | 0.406 | >0.05 | 0(0) | 0(0) | 1(1.7) | 1(2.8) | 0(0) | 0(0) | 1(3.7) | |
| 其他细菌 | 16(5.6) | 9(4.4) | 7(8.2) | 1.650 | >0.05 | 3(3.7) | 0(0) | 8(13.8) | 2(5.6) | 2(10.5) | 1(9.1) | 3(11.1) | |
| 合计 | 55(19.1) | 28(13.8) | 27(32.9) | 12.525 | <0.05 | 9(11.0) | 18(22.0) | 14(24.1) | 8(22.2) | 4(21.1) | 2(18.2) | 9(33.3) | |
| 真菌 | 36(12.5) | 23(11.3) | 13(15.3) | 0.861 | >0.05 | 4(4.9) | 16(19.5) | 8(13.8) | 4(11.1) | 2(10.5) | 1(9.1) | 3(11.1) | |
| 结核分枝杆菌 | 28(9.7) | 24(11.8) | 4(4.7) | 3.457 | >0.05 | 6(7.3) | 8(9.8) | 6(10.3) | 6(16.7) | 2(10.5) | 0(0) | 2(7.4) | |
| 支原体 | 16(5.6) | 11(5.4) | 5(5.9) | 0.025 | >0.05 | 2(2.4) | 3(3.7) | 5(8.6) | 2(5.6) | 2(10.5) | 2(18.2) | 1(3.7) | |
| 巨细胞病毒 | 14(4.9) | 6(3.0) | 8(9.4) | 5.400 | >0.05 | 3(3.7) | 5(6.1) | 2(3.4) | 4(11.1) | 0(0) | 0(0) | 0(0) | |
| EB病毒 | 13(4.5) | 12(5.9) | 1(1.2) | 3.116 | >0.05 | 1(1.2) | 3(3.7) | 3(5.2) | 5(13.9) | 1(5.3) | 0(0) | 0(0) | |
| 腺病毒 | 6(2.1) | 3(1.5) | 3(3.5) | 1.236 | >0.05 | 0(0) | 3(3.7) | 1(1.7) | 0(0) | 2(10.5) | 0(0) | 0(0) | |
注:三大病原:细菌、真菌、结核分枝杆菌 Three major pathogens:bacteria,fungi and Mycobacterium tuberculosis
总体而言,82例患儿检出病原依次为细菌(55/168例,32.7%)、真菌(36/168例,21.4%)、结核分枝杆菌(28/168例,16.7%),三者比较差异有统计学意义(P<0.05)。死亡组总体病原检出率、三大病原检出率、细菌检出率高于存活组,差异均有统计学意义(均P<0.05)。
82例患儿共培养出55株细菌,大多为革兰阴性菌。肺炎克雷伯菌共培养出10株,2株缺乏药敏资料,其余8株均为超广谱β-内酰胺酶(ESBL)阳性菌,对氨苄西林完全耐药,对复方磺胺甲 唑(SMZ)及三代头孢(头孢噻肟、头孢他啶)耐药率为87.5%;但对碳青霉烯类(美罗培南、亚胺培南)均敏感。5株大肠埃希菌中,1株缺乏药敏资料,余4株均为ESBL阳性菌,对氨苄西林、头孢噻肟完全耐药,对左氧氟沙星、SMZ耐药率为75.0%;同样对碳青霉烯类均敏感。洋葱伯克霍尔德菌共5株,其中4株对头孢他啶、SMZ敏感,1株对左氧氟沙星耐药;1株外院血培养提示洋葱伯克霍尔德菌,仅通过用药可知使用左氧氟沙星、万古霉素、四代头孢疗效均不明显。8株金黄色葡萄球菌仅2株有药敏结果,其中1株为耐甲氧西林金黄色葡萄球菌。
唑(SMZ)及三代头孢(头孢噻肟、头孢他啶)耐药率为87.5%;但对碳青霉烯类(美罗培南、亚胺培南)均敏感。5株大肠埃希菌中,1株缺乏药敏资料,余4株均为ESBL阳性菌,对氨苄西林、头孢噻肟完全耐药,对左氧氟沙星、SMZ耐药率为75.0%;同样对碳青霉烯类均敏感。洋葱伯克霍尔德菌共5株,其中4株对头孢他啶、SMZ敏感,1株对左氧氟沙星耐药;1株外院血培养提示洋葱伯克霍尔德菌,仅通过用药可知使用左氧氟沙星、万古霉素、四代头孢疗效均不明显。8株金黄色葡萄球菌仅2株有药敏结果,其中1株为耐甲氧西林金黄色葡萄球菌。
除易感染外,CGD患儿还可合并炎症并发症。纳入的82例患儿中17例(20.7%,存活组12例,死亡组5例)有不同程度的炎症表现,主要包括间质性肺病(ILD)或肺纤维化、炎症性肠病(IBD)、不完全川崎病三方面,且多系统炎症可并存,表现可相互重叠。4例合并ILD或肺纤维化;4例合并IBD;3例同时合并ILD和IBD;3例首发表现为不完全川崎病,其中1例合并ILD;另有2例表现为传染性单核细胞增多症;1例表现为白塞病。
分析这17例患儿的实验室检查结果,可发现11例(64.7%)患儿白细胞升高,14例(82.4%)伴贫血,12例(70.6%)伴血小板增多、C反应蛋白升高;其中11例患儿检测红细胞沉降率,均发现红细胞沉降率增快。在表现为传染性单核细胞增多症的2例患儿中,外周血可见异常淋巴细胞,但比例<0.1,其中1例血EBV-PCR阳性(1.9×109 copies/L),1例阴性。
8例胸部CT可见ILD表现。7例有IBD样表现,其中5例行肠镜后诊断为IBD。3例首发表现为不完全川崎病的患儿,心脏超声有不同程度异常:1例双侧冠状动脉扩张、左心室轻度增大;1例左侧冠状动脉轻度扩张、右侧冠状动脉正常高值;1例左侧冠状动脉正常高值。另有1例以"反复口腔溃疡7年"为主诉的患儿考虑白塞病,既往有过敏性紫癜样皮疹,尿血+++,但基因检测提示CYBB突变,诊断为CGD。
诊断前24例(29.7%)患儿接受过抗结核治疗,这部分患儿的确诊时间(43.5个月)晚于未接受抗结核治疗的患儿(7.4个月),差异有统计学意义(P<0.05)。
确诊后有58例(70.7%)患儿开始口服SMZ预防感染,平均用药36.8个月;1例因对SMZ过敏使用阿奇霉素,1例因葡萄糖-6-磷酸脱氢酶(G-6-PD)缺乏使用克林霉素。52例(63.4%)患儿开始应用伊曲康唑或伏立康唑预防真菌感染,平均用药34.3个月。5例患儿接受了数月γ-干扰素治疗,但因不良反应相继停用。30例(36.6%)患儿接受抗结核药物治疗。
在积极预防真菌感染下,患儿额外需要抗真菌治疗的比例由20.7%降至9.1%,但差异无统计学意义;抗细菌、抗结核类药物使用比例变化不大,分别维持在60%、30%左右。总体而言,患儿使用抗生素的比例:抗细菌类>抗结核类>抗真菌类。通过对比存活组和死亡组患儿使用特殊级抗生素(主要包括四代头孢菌素、β内酰胺酶抑制剂、碳青霉烯类和万古霉素)的情况,可发现死亡组患儿使用比例高于存活组,诊断时两者比例分别为70.4%、40.0%,差异有统计学意义(P<0.05);诊断后第1次住院两者比例为78.6%、29.5%,差异有统计学意义(P<0.05);第2次住院时两者比例为71.4%、24.1%,差异有统计学意义(P<0.05)。提示预后较差的患儿可能因感染较重,更加依赖于高级抗生素的治疗。但对于抗真菌类抗生素和抗结核治疗,2组差异无统计学意义。
82例患儿中32例(39.0%)接受了HSCT,其中25例(30.5%)CYBB基因突变患儿于重庆医科大学附属儿童医院接受移植,移植的平均年龄为40.2(8.5~117.4)个月,中位随访时间38.8个月。
重庆医科大学附属儿童医院移植患儿中14例行HLA相合无关供者(MUD)移植,采用白消安(Bu)+环磷酰胺(Cy)+抗胸腺免疫球蛋白(ATG)的减强度预处理(RIC)方案;11例行HLA相合相关供者(MRD)移植,其中6例全相合患儿采用氟达拉滨(Flu)+Bu+Cy预处理,3例半相合患儿采用Bu+Cy+ATG,早期移植的2例患儿采用了Bu+Cy的完全清髓预处理方案。24例移植成功,成功率为96.0%;除1例失败、1例嵌合度不详外,余23例(92.0%)患儿嵌合度均为100%。Ⅰ~Ⅱ级和Ⅲ~Ⅳ级急性GVHD的发生率分别为60.0%(15例)和4.0%(1例),主要为皮肤、消化道表现;4例患儿发生慢性GVHD,主要为肝脏GVHD。25例患儿移植后均接受丙种球蛋白支持治疗,输注丙种球蛋白平均时间8.5(0~19.0)月。13例(52.0%)患儿出现并发症或移植后再入院:因感染再入院13次,最主要的再入院原因为感染性腹泻或轮状病毒性肠炎(69.2%),其次为肺炎(30.8%);非感染再入院7次,原因包括:免疫性血小板减少(伴或不伴出血)(57.1%)、自身免疫性溶血性贫血(14.3%)等。
17例合并炎症并发症的患儿中,9例接受过抗炎治疗或使用免疫抑制剂/免疫调节剂。合并ILD患儿中4例使用小剂量阿奇霉素改善肺纤维化,疗效有限。IBD表现的患儿中,1例予地塞米松磷酸钠抗炎,3例予美沙拉嗪抗炎(1例合并使用英夫利昔单抗、1例合并使用干扰素)。考虑不完全川崎病的患儿中有1例应用大剂量丙种球蛋白冲击治疗及阿司匹林抗炎后冠状动脉情况好转。
死亡组患儿的平均年龄为37.6(2.0~190.3)个月。其中10例死于呼吸(呼吸-循环)衰竭;5例死于严重脓毒症(3例合并多器官功能衰竭);12例死亡原因不明(如惊厥发作、脓疱疮等)。死亡组检出病原以真菌、肺炎克雷伯菌、金黄色葡萄球菌、洋葱伯克霍尔德菌为主。
根据上述新生儿期有无症状、抗生素使用、移植情况等信息,绘制Kaplan-Meier生存分析曲线(图1),结果表明新生儿期即出现症状(P=0.002)、依赖于特殊级抗生素治疗(P=0.001)和未接受移植(P=0.000)的患儿生存率更低。

重庆医科大学附属儿童医院在既往研究中讨论了CGD的发病、感染部位、X连锁CGD(XL-CGD)与常染色体隐性遗传CGD(AR-CGD)的比较及CGD患儿的实验室检查特点;CGD患儿以反复发热(100.0%)、肺炎(92.1%)、脓肿(73.7%)、淋巴结炎(58.8%)、肝脾大(58.8%)、腹泻(45.6%)和败血症(30.7%)为主要表现[4] 。本研究作为补充,回顾性分析了82例CGD患儿的感染、炎症并发症的治疗情况。
结合既往研究,重庆医科大学附属儿童医院99.0%的CGD患儿起病年龄在3岁以内;本研究82例患儿平均起病年龄为3.7个月。CGD患儿多在5岁前确诊,及时诊断可减少并发症的发生、改善患儿预后[6,7];本组患儿中63例(76.8%)于3岁前确诊。新生儿期有症状的患儿确诊时间更早,且死亡组比例显著高于存活组;这可能与疾病特点及严重程度有关,提示临床表现出现早的患儿更早被诊断,但预后也更差。另一方面,诊断前接受过抗结核治疗患儿的确诊时间晚于未接受抗结核治疗的患儿,提示可能有部分患儿早期被误诊为结核感染给予抗结核治疗,从而延迟了确诊时间。患儿接种卡介苗后可引起了局灶性感染或播散性卡介苗病[8] ,中国大陆(上海)、中国香港、伊朗CGD患儿出现异常反应或发展为卡介苗病的比例分别为64%、47%、56%[9] 。本研究中患儿出现卡介苗接种反应的比例为58.6%,但这部分患儿接种反应的严重程度与生存情况无直接关联。
文献中已报道CGD患儿易感染过氧化物酶阳性的细菌和真菌(如金黄色葡萄球菌、曲霉菌、伯克霍尔德菌、黏质沙雷菌等)和一些不太常见的假单胞菌、念珠菌、镰刀菌属等[10,11],在一些流行地区,患儿易感沙门菌、结核分枝杆菌[12,13,14]。我国北方地区曲霉菌和结核分枝杆菌最常见;伊朗等中东地区以金黄色葡萄球菌、结核分枝杆菌、曲霉菌常见[15];美国则以金黄色葡萄球菌、黏质沙雷菌、洋葱伯克霍尔德菌、诺卡菌属常见[16] 。本研究患儿总体病原检出率较低,这可能与患儿长期预防性使用抗生素有关。在积极预防真菌感染下,真菌检出比例呈下降趋势,患儿额外需要抗真菌治疗的比例减少;但细菌、结核检出比例变化不大,抗细菌类抗生素的使用比例居高不下,这可能暴露了当前预防治疗仍有诸多不足。
死亡组细菌感染比例高,提示细菌感染对CGD患儿的预后影响更大。检出细菌以肺炎克雷伯菌、金黄色葡萄球菌、洋葱伯克霍尔德菌、大肠埃希菌为主,且耐药菌较多,如肺炎克雷伯菌、大肠埃希菌在现有的药敏资料中显示ESBL阳性比例高达100.0%。与此同时,死亡组使用特殊级抗生素比例更高,从侧面反映出一旦出现感染,感染仍重且治疗难度较大。以肺炎克雷伯菌为例,其为革兰阴性肠杆菌,有研究表明产ESBL、对碳青霉烯类耐药的细菌与ESBL阴性、不耐药的细菌相比,由于缺乏有效的治疗,可致更高的死亡率和病死率[17] 。本研究中洋葱伯克霍尔德菌占细菌类的第3位,在死亡前检出相对较多,提示洋葱伯克霍尔德菌感染可能与预后不良有关,这与此前的一项研究相符合[9] ,但例数较少。
炎症并发症是CGD患儿的另一重要表现。目前关于CGD合并ILD的报道较少[18] ,有学者提出使用肺功能和高分辨率胸部CT评估患儿肺部情况,暂无有效治疗方案,本组患儿仅使用小剂量阿奇霉素。IBD是CGD的常见并发症之一,常需联用小剂量激素及免疫抑制剂(如美沙拉嗪)抗炎,在严格的抗感染预防治疗下可使用甲氨蝶呤或巯嘌呤治疗反复发作患者[5],本研究纳入的患儿主要使用美沙拉嗪抗炎,有效性有待长期随访。CGD患儿可以不完全川崎病为首发表现,CGD和川崎病都与中性粒细胞功能有关,外周血可见中性粒细胞异常升高,故有报道称CGD患儿发生不完全川崎病可能与中性粒细胞过度活化有关[19]。总体而言,关于CGD患儿炎症并发症的发生机制、治疗措施还需进一步研究。
HSCT是根治CGD的方法之一[20,21],国外14岁以下CGD患儿经HSCT治疗后2年总体生存率达90.0%以上[22]。近年来减强度预处理方案的运用极大地减少了毒性作用和感染风险,纳入患儿移植成功率达96.0%,中位随访时间38.8个月,暂未出现死亡,这可能与我国移植起步较晚且采用减强度预处理有关,患儿移植后主要因肠道感染住院。然而目前由于供体不足、经济等原因病例数有限。
综上,CGD患儿临床表现出现越早、特殊级抗生素使用比例越高,提示病情越重,目前药物保守治疗仍有不足,耐药菌的产生更增加了治疗难度。纳入患儿的炎症并发症较少,可能与认识不足有关,相关领域的诊断和治疗仍有待加强。HSCT是目前CGD患儿的唯一治愈方法。
所有作者均声明不存在利益冲突

























