
对2019年1月安徽省儿童医院神经内科收治的1例4H综合征患儿的临床资料进行回顾性分析。患儿,男,2岁7个月,临床表现为运动及精神发育迟滞、步态不稳、牙齿发育异常;头颅磁共振成像显示脑白质发育异常;家系全外显子检测提示患儿存在POLR3A基因exon29:c.3858C>A及exon24:c.3226G>A复合杂合变异,为新的致病突变位点。提示对于早期存在发育落后、牙齿发育异常、脑白质异常的患儿需考虑4H综合征的可能。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
4H综合征(hypomyelination,hypodontia,hypogonadotropic,hypogonadism)是一种少见的遗传性脑白质病,其特征是髓鞘形成不良、牙发育不全和低促性腺激素性性腺功能减退,属于POLRⅢ相关的脑白质营养不良、最早由Timmons等[1]于2006年提出,报道脑白质髓鞘化不良、低促性腺激素性性腺功能减退、少牙畸形的患者,后逐渐发现同时可伴共济失调、白内障、小脑和胼胝体萎缩、椎体融合、周围神经髓鞘形成不良等其他临床表现。本研究分析比较POLR3A基因新生突变致4H综合征患儿的临床特征,扩展了4H综合征的基因型及表型。
患儿男,2岁7个月,2019年1月因"步态不稳6个月余"入住安徽省儿童医院神经内科。患儿近半年无诱因下走路时步态不稳、左右摇晃、容易跌倒,逐渐出现双上肢持物抖动,持物及取物时症状明显,追物欠灵敏,目前仍不能言语,自幼喂养困难,经常呕吐。入院前曾在外院考虑"发育迟滞"并给予康复治疗。父母非近亲婚配,母孕期正常,为第1胎,第1产,足月顺产,出生体质量3.2 kg,否认出生后窒息、病理性黄疸等特殊围生期疾病史。
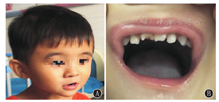
入院时查体:头围45 cm,身高80 cm,体质量9 kg(低于同年龄性别的第3百分位),神志清楚,精神可,反应一般,表情欠灵活,短人中,小下颌,上唇薄,牙齿不齐,出牙不全(图1),言语不能,双侧瞳孔等大等圆,对光反射灵敏,眼球活动自如,眼球震颤(+),颈软,心、肺、腹查体未见异常,行走不稳,步基增宽,指鼻试验(+),四肢肌力、肌张力正常,膝腱反射稍活跃,病理征阴性。
实验室检查:血常规、生化、心肌酶谱、血氨、丙酮酸、甲状腺功能未见异常;脑脊液常规、生化、培养、涂片均阴性;生长激素2.33 μg/L,总睾酮0.69 nmol/L,皮质醇83 nmol/L(正常值138~569 nmol/L);血尿遗传代谢性疾病筛查未见异常。头颅磁共振成像(MRI)检查示双侧大小脑半球白质区弥漫性异常信号,考虑遗传代谢性脑白质病(图2)。视频脑电图示背景活动偏慢,左侧额、颞区慢波发放。眼底检查提示双侧视神经有萎缩倾向。牙片提示牙列不齐及出牙不全。听力诱发电位双侧Ⅰ波潜伏期延长、双侧听性脑干反应(ABR)主观听阈轻度增高;视觉诱发电位示双侧P100潜伏期延长。
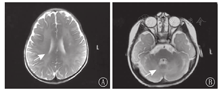
注:T2WI:T2加权像 T2WI:T2 weighted image
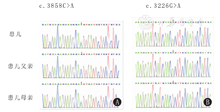
本研究获得患儿监护人知情同意,并通过安徽省儿童医院医学伦理委员会批准(批准文号:EYLL-2018-009)。分别采集患儿及其父母外周血2 mL,提取DNA,采用全外显子测序方法分析目的基因,经全外显子组测序及Sanger验证后发现患儿POLR3A基因(OMIM号606794)存在复合杂合突变(图3):c.3858C>A(p.His1286Gln)编码区第3858号核苷酸由C变为A,该变异导致第1 286号氨基酸由His变为Gln,患儿母亲携带相同的基因变异;同时c.3 226 G>A(p.Ala1076Thr)编码区第3 226号核苷酸由G变为A,该变异导致第1 076号氨基酸由Ala变为Thr,患儿父亲携带相同的杂合变异。以上均为错义突变,该变异在多个公共人群数据库(包括千人基因组、ExAC数据库等)均未见收录,多个蛋白功能预测软件(包括SIFT、PROVEAN、Polyphen2和Mutation Taster等)均预测这2个错义突变对蛋白功能有害。
目前随访近1年,院外一直给予B族维生素治疗,同时予康复功能锻炼,目前仍不能言语交流,走路需要扶行,复查头颅MRI示白质区无明显改善,发育测评明显落后于同龄儿。
2014年Wolf等[2]统计了105例经基因确诊的4H脑白质营养不良病例,发现其在临床表现的严重程度上呈很大不同,严重情况下患儿可出现不能正常行走,轻度至中度智力障碍,轻症者只表现出学习障碍和运动笨拙。该疾病主要在欧洲人群中报道,包括法裔加拿大人、高加索人、叙利亚人及印度人,中国家庭的报道病例偏少。
4H综合征主要表现出神经和非神经系统表现[2]。神经系统症状主要包括智力运动发育落后、共济失调、眼球震颤和肌张力障碍,头颅MRI检查提示髓鞘发育不良,视辐射、苍白球、齿状核、小脑萎缩和胼胝体变薄。非神经系统表现包括牙齿异常、视力改变及内分泌紊乱。牙齿主要表现为牙齿延迟、牙齿发育不全、牙齿减少及牙齿形态和排列异常。视觉主要表现为近视、白内障、视神经萎缩。内分泌主要表现在低促性腺激素性性腺功能减退伴青春期延迟或停止,身材矮小。通常发病较早的患儿表现出更多的是共济失调、眼球震颤、牙齿发育不良及脑白质营养不良,而性腺发育是否异常需要在青春期才能逐步被观察到,符合本研究患儿临床特点,同时也是4H综合征婴幼儿期需要注意的表现。
目前已认识到RNA聚合酶Ⅲ(RNA polymerase Ⅲ,RNA Pol Ⅲ)相关的脑白质营养不良涉及POLR3A、POLR3B、POLR1C和POLR3K(通过与POLR3B的相互作用)基因的突变,并且遗传方式为常染色体隐性遗传[3]。除4H综合征外,还包括其他表型,如共济失调、发育延迟、髓鞘形成减少型,震颤性共济失调伴中央低髓鞘化型,脑白质营养不良伴牙齿发育不良型及低髓鞘化小脑萎缩合并胼胝体萎缩型[4]。起病的年龄与疾病严重程度和表型相关,越早发病临床表现越严重,而POLR3A基因突变可导致更严重的临床表型[5]。这些基因负责编码RNA Pol Ⅲ,被认为是合成小RNA的关键,如5SrRNA、tRNAs、snRNA。其突变会导致细胞类型和生长状态异常,并可能影响细胞生长、分化和凋亡[6]。自2011年Bernard等[5]报道并提出4H综合征为POLR3A核苷酸变异引起,后逐渐扩展到POLR3A或POLR3B核苷酸变异均可引起4H综合征,并提出POLR3A核苷酸突变更常见、更频繁,同时4H综合征患者POLR3A或POLR3B基因新的不同核苷酸位点变异不断被发现,而对于本例POLR3A基因c.3858C>A/c.3226G>A在既往报道中未发现,故考虑为新发突变。在法国、加拿大,白种人和叙利亚人中发现了POLR3A基因的14个隐性突变[7],但对于中国人口中的病例仍不清楚。在本研究中,患儿以步态不稳来就诊,同时存在运动及精神发育延迟、牙齿发育异常及脑白质病变,随访半年脑白质病变范围未明显改善,符合其4H综合征的核心症状,进而完善基因进行验证后明确诊断。虽然文献提到该病可合并低促性腺激素性性腺功能减退,但考虑到患儿未到第二性征发育年龄,除测定皮质醇略低于正常水平外,内分泌科建议后期定期进行生长激素及促性腺激素释放激素激发试验并长期随访。本研究随访复查患儿MRI显示髓鞘未明显改善,提示MRI可识别早期不伴牙齿及内分泌异常的患儿,是诊断4H综合征的主要支持依据。对于患儿自幼吞咽困难,且经常呕吐的表现,考虑与小脑病变有关,并建议其监护人避免院外服用可能导致窒息的食物及多巴胺D2受体拮抗剂的药物(如甲氧氯普胺),因为这些药物可加重椎体外系症状。
尽管迄今为止尚无法治愈该病,但仍可对个体进行治疗,如改善癫痫发作、低促性腺激素性性腺功能减退、肌张力障碍和吞咽困难等表现,从而进一步改善患儿生活质量和预防并发症的出现。
所有作者均声明不存在利益冲突

























