了解河南省新生儿极长链酰基辅酶A脱氢酶缺乏症(VLCADD)的患病率、基因变异特点及预后。
采用串联质谱分析技术对2013年1月至2019年12月河南省867 103例新生儿行遗传代谢病筛查,对确诊VLCADD患儿及家系进行二代测序技术靶向测序及Sanger测序验证,分析其临床、生化改变及基因变异特点,进行饮食指导,随访观察其生长发育情况。
共确诊VLCADD新生儿6例,河南省新生儿VLCADD患病率为1/144 517。发现ACADVL基因变异11种,包括5种新发变异c.692-2_692-1delAG、c.753-23_753-22del、c.960delG、c.1361A>G、c.1955C>T。对确诊新生儿给予高碳水化合物、低脂饮食,随访8~56个月,除2例患儿死亡外,其余患儿预后良好。
河南省新生儿VLCADD患病率为1/144 517。本研究结果丰富了ACADVL基因突变谱,为VLCADD筛查和诊断提供了重要依据。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
极长链酰基辅酶A脱氢酶缺乏症(VLCADD,OMIM#201475)是由于细胞线粒体内脂肪酸β氧化中的关键酶极长链酰基辅酶A脱氢酶(VLCAD)ACADVL(609575)基因变异导致的一种常染色体隐性遗传的脂肪酸代谢障碍疾病,是婴儿期潜在猝死性疾病之一[1,2]。VLCADD临床表型有异质性,表现为不同的严重程度和发病年龄,可发生在新生儿期、婴儿期、儿童期或成年期。临床上有3种表型:新生儿早发型又称心肌病型,以心肌病、脑病、Reye综合征为主要表现,此型发病凶险,患儿病死率高;肝型多在婴儿晚期或幼儿期起病,以肝脏病变及低酮症性低血糖为主要表现;肌病型多在青少年或成人期起病,以间歇性横纹肌溶解及发作性肌肉病为主要表现[3]。该病起病隐匿,首次发病的病死率及神经系统后遗症发生率高,其患病率、基因型存在明显种族差异[4]。目前国内尚未见VLCADD流行病学、基因型及远期预后的报道。本研究基于河南省新生儿疾病筛查中心2013年1月至2019年12月的高效液相色谱串联质谱(high-performance liquid chromatographytandem mass spectrometry,HPLC-MS/MS)筛查资料库,分析河南省新生儿VLCADD患病率,探讨其表型、基因型特征及预后,为VLCADD防控提供循证依据。
2013年1月至2019年12月来自河南省18个地市的在河南省新生儿疾病筛查中心进行遗传代谢病筛查的867 103例新生儿。样本的采集和检测均在监护人签署知情同意书的情况下完成,本研究通过郑州大学第三附属医院医学伦理委员会批准[批准文号:(2019)医伦审第67号]。
采集新生儿出生3~7 d足底血,滴于专用采血滤纸上晾干后送检。采用waters液相色谱-串联质谱联用系统,芬兰PerkinElmer公司的非衍生化试剂盒(串联质谱)检测干血滤纸片中的氨基酸及酰基肉碱。初筛阳性者进行复查,仍阳性者进一步行基因突变分析。生化诊断:(1)筛查可疑的判断参照文献[1],多种长链酰基肉碱水平增高如C14∶1、C14、C14∶2、C16、C18,其中肉豆蔻烯酰肉碱C14∶1高于正常2倍,正常参考(0.01~0.30 μmol/L),或C14∶1合并C14∶1/C10(肉豆蔻烯酰肉碱/葵酰基肉碱)增高作为VLCADD可疑病例。(2)常规实验室检查,包括血尿常规、血糖、血氨、血乳酸、肝肾功能、肌酸激酶(CK)、心电图、心脏彩超等。
采集6例患儿及其父母外周血各2 mL(乙二胺四乙酸抗凝),采用二代测序技术靶向测序并进行Sanger测序验证。基因测序结果与ESP6500数据库、千人基因组数据库、人类基因变异数据库(HGMD数据库)、Clinvar数据库等公共数据库进行比对,按照美国医学遗传学与基因组学学会(ACMG)关于基因变异位点的解读原则进行解读,获得致病性变异位点信息[5]。
对确诊新生儿避免空腹、感染和疲劳,给予高碳水化合物、富含中链三酰甘油(MCT)的特殊奶粉(Monogen,荷兰纽迪西亚公司),低脂饮食。治疗初期每2周门诊随访1次,血肉碱正常且稳定后2~3个月随访复查1次,治疗期间定期复查串联质谱。随访项目包括:体格检查、血糖、血氨、肝功能、CK、血红蛋白、心脏彩超等,评估生长、智力及运动发育情况,随访时间8~56个月。同时做好家长宣教工作,嘱其长期规范饮食干预,避免饥饿、加强护理、积极预防和控制感染等。
VLCADD初筛阳性新生儿264例,召回261例,召回率98.86%。确诊6例VLCADD,河南地区VLCADD患病率为1/144 517。6例患儿均显示有C14∶1肉碱水平升高(表1)。确诊患儿均为男性足月儿,父母非近亲结婚,无遗传病家族史。
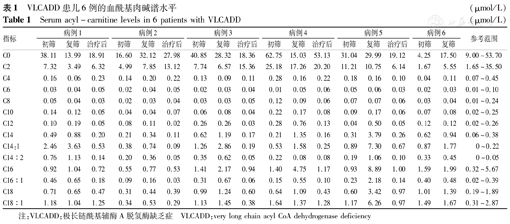
VLCADD患儿6例的血酰基肉碱谱水平(μmol/L)
Serum acyl-carnitine levels in 6 patients with VLCADD (μmol/L)
VLCADD患儿6例的血酰基肉碱谱水平(μmol/L)
Serum acyl-carnitine levels in 6 patients with VLCADD (μmol/L)
| 指标 | 病例1 | 病例2 | 病例3 | 病例4 | 病例5 | 病例6 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 初筛 | 复筛 | 治疗后 | 初筛 | 复筛 | 治疗后 | 初筛 | 复筛 | 治疗后 | 初筛 | 复筛 | 治疗后 | 初筛 | 复筛 | 治疗后 | 初筛 | 复筛 | 参考范围 | |
| C0 | 38.11 | 13.99 | 18.91 | 16.60 | 32.12 | 27.98 | 40.85 | 28.32 | 18.36 | 62.75 | 15.03 | 53.13 | 31.04 | 29.99 | 19.12 | 4.25 | 17.50 | 9.00~53.70 |
| C2 | 7.32 | 3.49 | 6.32 | 4.99 | 7.85 | 13.12 | 7.74 | 6.57 | 15.36 | 25.18 | 17.26 | 20.20 | 11.21 | 10.75 | 6.14 | 1.67 | 5.55 | 1.65~35.50 |
| C4 | 0.16 | 0.06 | 0.23 | 0.14 | 0.20 | 0.22 | 0.13 | 0.09 | 0.11 | 0.28 | 0.16 | 0.22 | 0.18 | 0.16 | 0.10 | 0.04 | 0.11 | 0.07~0.45 |
| C6 | 0.03 | 0.04 | 0.05 | 0.02 | 0.04 | 0.05 | 0.02 | 0.03 | 0.04 | 0.01 | 0.05 | 0.06 | 0.05 | 0.06 | 0.03 | 0.02 | 0.03 | 0.01~0.10 |
| C8 | 0.05 | 0.04 | 0.03 | 0.02 | 0.03 | 0.04 | 0.03 | 0.03 | 0.05 | 0.12 | 0.09 | 0.06 | 0.07 | 0.07 | 0.06 | 0.03 | 0.04 | 0.01~0.24 |
| C10 | 0.14 | 0.12 | 0.05 | 0.04 | 0.04 | 0.07 | 0.06 | 0.08 | 0.04 | 0.22 | 0.17 | 0.08 | 0.09 | 0.17 | 0.06 | 0.07 | 0.08 | 0.02~0.25 |
| C12 | 0.10 | 0.19 | 0.05 | 0.08 | 0.11 | 0.02 | 0.26 | 0.26 | 0.03 | 0.28 | 0.76 | 0.13 | 0.04 | 0.50 | 0.05 | 0.12 | 0.12 | 0.02~0.26 |
| C14 | 0.49 | 0.88 | 0.20 | 0.21 | 0.34 | 0.11 | 0.62 | 1.19 | 0.17 | 0.21 | 1.35 | 0.16 | 0.31 | 3.79 | 0.26 | 0.62 | 0.94 | 0.06~0.38 |
| C14:1 | 2.46 | 3.63 | 0.53 | 0.38 | 0.74 | 0.09 | 1.26 | 2.86 | 0.19 | 0.53 | 1.58 | 0.25 | 0.89 | 7.30 | 0.67 | 0.87 | 1.77 | 0~0.22 |
| C14∶2 | 0.76 | 1.13 | 0.14 | 0.20 | 0.36 | 0.05 | 0.35 | 0.62 | 0.05 | 0.22 | 0.08 | 0.08 | 0.19 | 1.06 | 0.10 | 0.33 | 0.45 | 0~0.05 |
| C16 | 0.92 | 1.04 | 0.72 | 0.55 | 0.77 | 0.53 | 1.41 | 2.17 | 0.94 | 1.40 | 4.75 | 1.17 | 0.93 | 8.89 | 1.00 | 1.59 | 1.99 | 0.32~5.67 |
| C16∶1 | 0.46 | 0.65 | 0.18 | 0.09 | 0.16 | 0.03 | 0.31 | 0.67 | 0.06 | 0.15 | 0.55 | 0.10 | 0.23 | 2.18 | 0.14 | 0.40 | 0.48 | 0.02~0.39 |
| C18 | 0.71 | 0.65 | 0.47 | 0.31 | 0.44 | 0.39 | 0.99 | 1.24 | 0.60 | 0.64 | 1.09 | 0.43 | 0.60 | 3.42 | 0.97 | 1.01 | 1.39 | 0.19~1.89 |
| C18∶1 | 1.18 | 1.04 | 1.25 | 0.34 | 0.53 | 0.29 | 1.13 | 1.45 | 0.38 | 1.64 | 1.37 | 1.28 | 1.17 | 6.26 | 0.97 | 1.49 | 1.67 | 0.31~2.87 |
注:VLCADD:极长链酰基辅酶A脱氢酶缺乏症 VLCADD:very long chain acyl CoA dehydrogenase deficiency
6例患儿常规实验室检查结果及随访见表2。有5例患儿确诊时均轻度肝功能异常,确诊后开始饮食治疗,给予富含MCT的特殊奶粉,治疗2周后血酰基肉碱谱水平较前明显下降,见表1,表2。病例1确诊后未规律饮食控制,7月龄时因不明原因呕吐住院发现肝大,谷丙转氨酶、谷草转氨酶、CK、C14和C14∶1肉碱的水平明显增高,规律治疗后临床症状显著改善,指标恢复正常,目前3岁8个月,生长发育基本正常;病例2确诊后未饮食控制,在8月龄时因"肺炎"住院超声心动图检查发现心包积液,心室增厚,左心收缩功能低下,谷丙转氨酶、谷草转氨酶、CK、C14和C14∶1肉碱的水平明显增高,给予富含MCT的特殊奶粉治疗后临床症状有显著改善,指标恢复正常,目前1岁8个月,生长发育基本正常;病例3和4确诊后一直无症状,1月龄开始富含MCT酸奶粉喂养,规律治疗,目前分别为2岁和8个月,生长发育基本正常;病例5系第3胎,第3产,其2个姐姐均在出生20 d内不明原因夭折,虽出生6 d确诊后给予富含MCT的特殊奶粉,6月龄时腹泻后猝死;病例6系第2胎,第2产,试管婴儿,出生10 d因"新生儿肺炎,低酮症性低血糖"夭折。
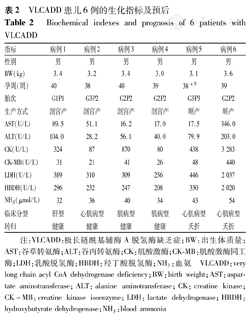
VLCADD患儿6例的生化指标及预后
Biochemical indexes and prognosis of 6 patients with VLCADD
VLCADD患儿6例的生化指标及预后
Biochemical indexes and prognosis of 6 patients with VLCADD
| 指标 | 病例1 | 病例2 | 病例3 | 病例4 | 病例5 | 病例6 |
|---|---|---|---|---|---|---|
| 性别 | 男 | 男 | 男 | 男 | 男 | 男 |
| BW(kg) | 3.4 | 3.2 | 3.4 | 3.0 | 3.1 | 3.6 |
| 孕周(周) | 40 | 38 | 40 | 39 | 38+5 | 39 |
| 胎次 | G1P1 | G3P2 | G2P2 | G2P2 | G3P3 | G2P2 |
| 生产方式 | 剖宫产 | 剖宫产 | 剖宫产 | 剖宫产 | 顺产 | 顺产 |
| AST(U/L) | 89.5 | 51.1 | 16.2 | 17.0 | 17.5 | 346.0 |
| ALT(U/L) | 104.0 | 28.2 | 56.1 | 40.0 | 79.9 | 203.0 |
| CK(U/L) | 324 | 87 | 870 | 80 | 438 | 3 283 |
| CK-MB(U/L) | 31 | 21 | 41 | 26 | 48 | 440 |
| LDH(U/L) | 389 | 310 | 309 | 256 | 446 | 2 037 |
| HBDH(U/L) | 296 | 232 | 247 | 208 | 330 | 2 020 |
| NH3(μmol/L) | 32 | 36 | 40 | 34 | 43 | 54 |
| 临床分型 | 肝型 | 心肌病型 | 肌病型 | 肌病型 | 心肌病型 | 心肌病型 |
| 转归 | 健康 | 健康 | 健康 | 健康 | 夭折 | 夭折 |
注:VLCADD:极长链酰基辅酶A脱氢酶缺乏症;BW:出生体质量;AST:谷草转氨酶;ALT:谷丙转氨酶;CK:肌酸激酶;CK-MB:肌酸激酶同工酶;LDH:乳酸脱氢酶;HBDH:羟丁酸脱氢酶;NH3:血氨 VLCADD:very long chain acyl CoA dehydrogenase deficiency;BW:birth weight;AST:aspartate aminotransferase;ALT:alanine aminotransferase;CK:creatine kinase;CK-MB:creatine kinase isoenzyme;LDH:lactate dehydrogenase;HBDH:hydroxybutyrate dehydrogenase;NH3:blood ammonia
6例患儿ACADVL基因2个等位基因均检测到变异,变异分别遗传自父母,共检出11种变异,包括6种错义变异,1种无义变异,2种剪切变异,1种移码变异和1种框移变异;其中c.65C>A、c.298_299del、c.553G>A、c.1226C>T、c.1276G> A、c.1345G>C为已知变异,c.692-2_692-1delAG、c.753-23_753-22del、c.960delG、c.1361A>G、c.1955C>T为新发变异(图1、表3),在HGMD数据库、Clinvar数据库等数据库中未发现这些位点的致病性的相关报道。本研究有2例患儿均携带有1个无义变异,目前临床表现基本正常;病例5虽携带了2个错义变异,出生6 d得以确诊并给予积极治疗,6月龄死于急性感染;病例6携带了1个错义突变和1个框移变异,出生10 d因"新生儿肺炎,低酮症性低血糖"夭折。患儿基因突变位点分散,未能发现表型与基因型的相关性。
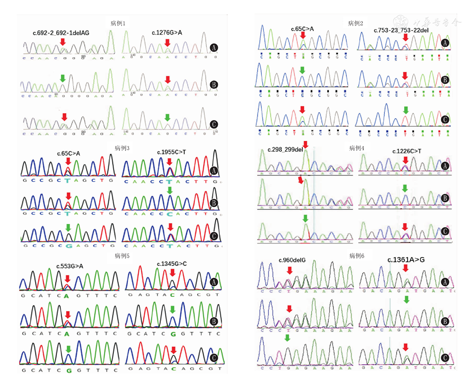
注:VLCADD:极长链酰基辅酶A脱氢酶缺乏症 VLCADD :very long chain acyl CoA dehydrogenase deficiency
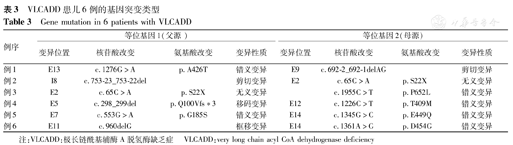
VLCADD患儿6例的基因突变类型
Gene mutation in 6 patients with VLCADD
VLCADD患儿6例的基因突变类型
Gene mutation in 6 patients with VLCADD
| 例序 | 等位基因1(父源) | 等位基因2(母源) | ||||||
|---|---|---|---|---|---|---|---|---|
| 变异位置 | 核苷酸改变 | 氨基酸改变 | 变异性质 | 变异位置 | 核苷酸改变 | 氨基酸改变 | 变异性质 | |
| 例1 | E13 | c.1276G>A | p.A426T | 错义变异 | E9 | c.692-2_692-1delAG | 剪切变异 | |
| 例2 | I8 | c.753-23_753-22del | 剪切变异 | E2 | c.65C>A | p.S22X | 无义变异 | |
| 例3 | E2 | c.65C>A | p.S22X | 无义变异 | c.1955C>T | p.P652L | 错义变异 | |
| 例4 | E5 | c.298_299del | p.Q100Vfs*3 | 移码变异 | E12 | c.1226C>T | p.T409M | 错义变异 |
| 例5 | E7 | c.553G>A | p.G185S | 错义变异 | E14 | c.1345G>C | p.E449Q | 错义变异 |
| 例6 | E11 | c.960delG | 框移变异 | E14 | c.1361A>G | p.D454G | 错义变异 | |
注:VLCADD:极长链酰基辅酶A脱氢酶缺乏症 VLCADD:very long chain acyl CoA dehydrogenase deficiency
VLCADD是一种常染色体隐性遗传的脂肪酸代谢障碍疾病,经串联质谱检测血酰基肉碱谱发现C14∶1特异性升高可诊断VLCADD疾病,通过ACADVL基因分析可进一步明确诊断。世界各国VLCADD患病率均较低,新生儿患病率为1/100 000~1/25 000,沙特阿拉伯为1/37 000,美国为1/63 481,日本为1/93 000[6]。我国尚未见全国性患病率的统计数据,苏州患病率约1/70 424[7],泉州约为1/91 136[8],湖南省约为1/188 394[9],浙江省约为1/236 665[10],本研究共筛查河南省新生儿867 103例,VLCADD初筛阳性新生儿264例,召回261例,召回率98.86%,确诊6例VLCADD,河南地区VLCADD患病率为1/144 517,与苏州、浙江等地相比有差异,可能与地域差异有关。
VLCAD作为线粒体脂肪酸β氧化过程关键酶,缺陷将导致体内长链脂肪酸不能氧化供能,蓄积在肝脏、心肌、骨骼肌、皮肤成纤维细胞内产生毒性作用,引起相应的临床症状和体征。通过新生儿筛查及早诊断并干预治疗能有效提高VLCADD患儿的生存率,改善预后。Pena等[11]报道了52例新生儿筛查确诊VLCADD患儿随访研究,大部分无临床症状。Rovelli等[12]对新生儿筛查确诊的24例VLCADD进行随访,虽然85%的VLCADD患者出现低血糖、纳差、嗜睡、肝功能异常、运动不耐受等症状,但早期给予干预治疗,患儿发育均基本正常。但也有研究发现新生儿筛查几乎不能改变重型患儿的预后,远期影响尚需进一步观察[13]。本研究中,心肌病型3例(50%)发病早,进展迅速,其中2例死亡。1例肝型患儿,未规律低脂饮食,7月龄时因不明原因呕吐住院发现肝大,谷丙转氨酶、谷草转氨酶、CK、C14和C14∶1肉碱的水平明显增高,规律治疗后临床症状有显著改善,指标恢复正常。2例肌型患儿目前均无明显症状,CK轻度增高,无肌溶解症状。所有存活患儿生长发育水平经身高体质量评估、发育商筛查,均提示基本正常。该研究提示VLCADD新生儿筛查早期诊治、规范随访对降低新生儿VLCADD的病死率意义重大。
VLCADD的致病基因ACADVL位于染色体17p13.1,长约5.4 kb,含20个外显子,编码655个氨基酸前体蛋白,位于线粒体内膜属于脂酰辅酶A脱氢酶(acyl-coenzyme adehydragenase,ACAD)家族成员之一。截至2020年8月,全球已报道了ACADVL基因332种变异(HGMD http://www.hgmd,cf.ac.uk/ac/index.php),以错义变异为主。ACADVL的变异也有一定的地域特征,c.848T>C致病性突变是欧美最常见的变异,中东人群中常携带c.65C>A变异,c.1349G>A是亚洲及中国人群中相对最常见的变异[14,15]。本研究共检出11种变异,异质性高,仅c.65C>A在2例患者检出,未检测到c.848T>C和c.1349G>A,也未见热点变异。本研究发现了5种ACADVL基因的新变异:c.692-2_692-1delAG,c.753-23_753-22del、c.960delG、c.1361A>G、c.1955C>T,通过VLCAD肽链的氨基酸序列,影响酶蛋白的一级结构,导致相应蛋白质功能的异常或丧失。新变异的致病性质需通过体外表达研究,以证实其对VLCAD功能的影响。新变异的发现,也进一步丰富了人类基因突变数据库。6例患儿共检出11种变异,其中错义变异6种,6例患儿携带6种不同的基因型,均为复合杂合变异,与国内外报道一致[15,16]。有研究显示VLCADD患者基因型和表型有一定相关性,通常携带2个无义变异(如终止、移码和剪切变异)的患儿临床表现较重且出生早期发病,而携带有至少1个错义变异的患儿则临床症状相对较轻且发病较晚[17]。本研究中有2例患儿均携带有1个无义变异,目前临床表现基本正常;病例5虽携带了2个错义变异,出生6 d得以确诊并给予积极治疗,6月龄死于急性感染;病例6携带了1个错义突变和1个框移变异,出生10 d因"新生儿肺炎,低酮症性低血糖"夭折,本研究并未发现基因型和表型有一定相关性,可能与确诊患儿例数较少,患儿基因突变位点分散、患儿均通过新生儿疾病筛查得以早期诊断和治疗有关。
研究显示,串联质谱新生儿筛查对新生儿VLCADD早期诊断和治疗有重要意义。VLCADD早期诊治、规范随访可改善患儿的预后。本研究采用二代测序技术对新生儿筛查发现的VLCADD患者进行了基因检测,从基因水平证实了临床诊断,也为VLCADD的遗传咨询、产前诊断及胚胎植入前遗传学诊断提供了重要信息。
所有作者均声明不存在利益冲突


























