糖皮质激素(GCs)应用于临床已有60余年的历史,临床上常用于治疗免疫性疾病和过敏性疾病,尤其近年来,随着免疫性及炎症性疾病发病率的增长,GCs类药物的临床应用也呈逐年上升趋势。因此,GCs的不良反应问题也愈发受到关注。剖析GCs作用机制,找到新的糖皮质激素受体(GR)配体,使其既有抗炎及免疫抑制作用,且不伴不良反应是医学领域亟待解决的重要问题。现就GR配体的研究进展作一综述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
糖皮质激素(GCs)是一类主要由肾上腺皮质分泌或人工合成的甾体激素[1]。GCs应用于临床已有60余年的历史,其在不同基础生命进程中均发挥了重要作用,是人体内必不可少的激素之一。内环境稳态、细胞增殖、分化、复制、炎症,乃至认知和心理健康[2]均与GCs息息相关。GCs具有抗炎、抑制免疫、抗休克等作用,是临床上治疗诸多免疫性疾病、炎症性疾病和过敏性疾病的首选药物。有调查结果显示,全世界约5‰的人口需常年使用GCs[3]。现就GCs及其受体工作原理和多方向发展方面,对糖皮质激素受体(GR)配体的研究进展作一综述。
GCs是亲脂性分子,可自由通过细胞膜扩散,与GR结合发挥作用。GR是由NR3C1基因编码的转录因子[4],主要存在于复杂的细胞质中,与热休克蛋白、亲免蛋白及其他防止其降解的因子共存,这些因子也可增强GR与其配体的亲和力。在与配体结合后,GR会产生形态变化,这导致其与分子伴侣复合体部分分离及核转移[5](图1)。在细胞核中,GR与DNA及其他蛋白质也存在交互作用[6]。
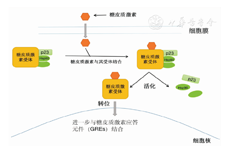
注:p23与Hsp90均为伴侣蛋白
p23 and Hsp90 are chaperones
GCs的释放受时间节律及应激反馈的调控[7]。疾病通常会扰乱其昼夜节律。目前研究证实,类风湿性关节炎(RA)的临床症状在晨起时最严重,归因于促炎细胞因子在睡眠时浓度较高[8]。因此,在睡前服用延迟释放的泼尼松,其疗效远胜于服用立即释放的泼尼松。由此可见,在不恰当的时间摄入GCs可能是GCs治疗失败的原因之一。
此外,在炎症进程中,GCs可通过抑制促炎信号通路及作用于炎症细胞来促进炎症反应的结束。全基因组研究表明,GR以特定的模式发挥作用[9]。根据其作用的细胞类型及其工作的生理环境的不同,GR可调节许多生理过程,而这些生理反应可能是治疗需求,也可能是所谓的"不良反应"。正如与GCs广泛的临床用途相对应的是其繁多的不良反应,如骨质疏松症、高血糖、胰岛素抵抗、干扰脂肪沉积、高血压和肌肉萎缩等,而临床所需要的正是分析GCs化学结构、药代动力学、分布等,多方寻找更合适的GR配体,在保证治疗效果的前提下尽量减少不良反应,甚至消除不良反应。
近几十年的研究普遍认为,是二聚体介导的转录活化(TA)过程诱导了GCs疗法的不良反应[10],TA即GR二聚体与GCs反应元件(GREs)的结合直接增强了基因的表达,该机制诱导的基因在糖合成和脂肪代谢中发挥作用[11],如由此类基因编码的肝脏中的葡萄糖6磷酸酶和磷酸烯醇丙酮酸羧激酶及脂肪细胞中的硬脂酰辅酶A去饱和酶1~3。而激素的抗炎作用通常被认为与单体介导的转录抑制有关[12],单体通过关键的炎症调节因子,如转录激活因子AP-1、核因子激活的B细胞κ-轻链增强蛋白复合物核因子-κB(NF-κB)等介导转录抑制过程。
通过对小鼠模型的研究,已发现一些由TA介导的不良反应,如高血糖和腹部肥胖等[13]。其他不良反应,如骨质疏松症,在长期使用GCs喂养的小鼠模型中,同程度地表现在了被抑制GR二聚体化小鼠模型和未处理小鼠模型上。此外,最近的研究表明,在一些疾病中,GR二聚体化的抗炎作用是必不可少的[14]。被抑制GR二聚体化的小鼠模型更易感急性炎症。而在更多慢性炎症的模型中,如刺激性皮肤炎症,抑制GR二聚体化小鼠的炎症与未处理小鼠相似,二者接受同一剂量GCs的治疗效果是一致的[15]。
因此,想要得到选择性抑制GR二聚体化治疗慢性炎症疾病的GR单体的配体,就意味着在功能上将TA与转录抑制分离。现有的几种选择性GR激动剂(SEGRAs)[16],如RU24782,RU24858及RU40066,具有与泼尼松龙一样强大的抗炎能力。然而,这些合成类固醇仍无法避免引起类似于GCs诱导的不良反应[17],还需扩大寻找的族系目标,从甾族的谱系扩大到非甾族。
化合物A(CpdA)是一种植物来源的GR配体,包含类似RA滑膜细胞的成分。从生物活性角度来看,CpdA类似传统的GCs,但CpdA减少了关节炎和神经炎症模型的炎症反应,同时并未诱导高血糖和高胰岛素血症的发生[18]。然而其不稳定性使其无法作为药物应用。所有与其相关的分离出的配体,仅有下面2种已进入临床阶段:Mapracorat已作为一种混悬型滴眼液局部用药用于治疗过敏性结膜炎、白内障术后疼痛及特应性皮炎[19];Fosdagrocorat已作为RA治疗药物进入Ⅱ期实验阶段,其效果胜于泼尼松龙且更不易引起GCs相关不良反应[20]。对于这些新化学物质长期使用的安全性和效果的深入研究是当务之急。
鉴于GCs的不良反应呈剂量依赖性,剂量减少时不良反应通常也随之减少。因此局部给药的GCs(如乳霜、鼻喷剂、吸入剂等)应用于许多炎症疾病的治疗中,可降低其进入循环系统的剂量。如吸入性激素到达肺部,可抑制气道炎症。但对于传统有效的GR激动剂来说,肺部给药不是其最佳药代动力学上的给药途径,GCs会迅速从肺部吸收到血液。为克服这个问题,将药物进行聚乙二醇化,形成一个大的亲水性前体药物,由此得到新的GR配体可使药物更多地留在肺组织中,从而降低不良反应[21]。
另一种方式是通过提升GR配体的亲脂性来提高疗效。丙酸氟替卡松是一种高亲脂性GR配体,可轻易穿透细胞膜的脂质双层膜。因此,丙酸氟替卡松作为吸入性药物非常恰当,可迅速被肺组织吸收,在肺部停留更长时间[22]。此外,脂质结合是一种GCs和组织内脂肪酸发生的化学反应,由此可产生一批储存的GCs,该反应是可逆的,可缓慢释放GCs,延长其作用时间,因此,可借此找到能被储存更多、更久的新GR配体,不必频繁服用药物[23],从而带来更少的不良反应。此外,其他通过修饰GCs理化性质得到的新GR配体也很有前景,如通过增加GCs的关键酯键使其只能局部用药,一旦进入血液循环,就可通过血液中的酯酶使药物失活,这个策略对于局部用药尤其有效,特别是肺部给药[24]。最近的研究还描述了一类拥有与GR结合更加紧密的共价键的GR配体,这样的配体能够更强烈地刺激GR,即使更小剂量也能达到抗炎效果,这也是局部应用GCs更可取的发展方向[25]。
减少不良反应的另一种方法是靶向治疗的GR配体。如使用一个抗体结合药物或多肽结合药物可精确给药至目标部位。地塞米松和CD163抗体的结合药物已被证实可精准针对活化巨噬细胞起效,无论体外或体内环境,该结合药物可减少活化巨噬细胞释放细胞因子,其体内效力是单纯地塞米松的50倍以上[26]。此外,胰高血糖素类似物多肽-1(GLP1)和地塞米松的结合物可特异性地将地塞米松传递至表达GLP1受体的细胞,目前正在实验用于代谢炎症和肥胖症的治疗[27]。GLP1-地塞米松可在治疗下丘脑和系统性炎症的同时改善机体葡萄糖耐量和胰岛素敏感性,且仅有最小程度的不良反应。基于这些成功案例,其他的抗体或多肽与GCs结合得到的GR新配体前景可期。
此外,将GCs封装处理得到的GR配体是尽可能降低GCs全身浓度的另一个选择。如脂质体目前已发展至可专门将GCs给药到RA的炎症关节、克罗恩病的炎症节段、结肠炎等[28]。因为脂质体更强大的渗透性和留存能力,可积蓄在有血管行走的组织中,如炎症区域和肿瘤部位,所以可最大程度上降低药物全身浓度。这就意味着药物在病变部位浓度的增加和起效时间的延长,在降低不良反应的同时提升治疗效率。目前已有完成的二期临床试验验证了用脂质体泼尼松治疗RA的安全性,且对比单纯泼尼松龙治疗有显著增高的疗效[28]。
另一个避免GCs相关不良反应的方法为刺激GR和PPARs之间的组织因子。PPARs是核激素受体家族中的配体激活受体,在不同的物种中已发现3种亚型,控制许多细胞内代谢过程,属配体诱导核受体。GR和PPARs均为细胞核受体成员,在许多组织中均有重叠和互补作用,控制参与维持血糖正常的关键基因,合作支持空腹时脂肪酸的β氧化,刺激免疫抑制[29]。PPARα和PPARγ是PPARs家族中2个很有研究前景的亚型。假设将GR和PPARα连接在一起,那么它们与GCs结合之后产生的抗炎效果可能会叠加,同时不良反应却并不会增加[30]。
GR和PPARα的功能性信号交叉揭示了它们各自配体受体的同步触发,同时影响GCs控制的2个相反方向的基因程序。联合的激动剂治疗理论上可得到很好疗效,因为可在增强对细胞因子抗炎作用的同时,避免GCs诱导的高血糖。这个假设已被一个高脂饮食的动物实验证实[31]。除控制血糖外,PPARα也可通过刺激脂肪酸的β氧化降低血脂水平,同时联合的GCs和PPARα激动剂共同活化参与脂质代谢的基因[32]。
PPARα和GR激动剂的联合药物不仅可提高抗炎效果、控制慢性炎症疾病中GCs相关的不良反应,还可用于溶血和遗传性骨髓衰竭疾病的治疗。动物研究表明,PPARα激动剂可能提高GCs治疗促红细胞生成素耐药的疗效[33]。通过对比敲除PPARα小鼠和普通小鼠,还可得出PPARα可介导GCs产生对于实验性脊髓损伤继发损伤的更好的抗炎效果[34]。
与GR和PPARα相似,PPARγ表现出的抗炎作用是通过对促炎的转移因子,如NF-κB、AP-1和信号转导与转录激活因子(STATs)等的干扰来实现的[34]。除基因抑制机制外,PPARγ进一步促使巨噬细胞向抗炎和促进内环境稳态的M2型极化[35]。目前已发现PPARγ配体对于消化道、肾缺血和再灌注损伤[35]及出血性休克的实验模型均有有益作用。此外,一个使用罗格列酮治疗溃疡性结肠炎的临床实验证实了PPARγ配体对炎症性肠病的治疗潜力[36]。
PPARγ和GR以及其结合物被证实参与减轻格列酮介导的胃溃疡相关炎症反应[37]。一些观点认为GR-PPAR信息交互对于一些使用GCs局部治疗的皮肤病有重要作用,如在特应性皮炎治疗中,与局部GCs用药一起联用PPARα或PPARγ的激动剂治疗可保持角化细胞分化,维持屏障内稳态[38]。
上述关于GR和PPARs的研究证实了联合类固醇激动剂治疗在多数疾病中(至少在慢性炎症疾病中)对比单独类固醇疗法的潜在优势。接下来的研究将是在体内环境中模拟类似情况行进一步验证。总的来说,多数GR是否具有抗炎活性取决于环境,如配体的类型、疾病或组织。结合2个或更多核受体特异配体,研究其抗炎能力和不良反应可能是新疗法的研究方向。
综上所述,区别于没有目的地寻找替代GCs的药物,从GCs与GR作用机制和过程出发,寻找GR新配体从而达到规避不良反应的效果显然具有很大可行性。无论是基于GCs的二聚体及单体理论、对GCs化学结构的修饰及靶向治疗或者是GR与PPARs的联合应用,这些途径都获得了许多新的灵感,和多种即将进入或正处于临床试验阶段的新治疗方法。随着学者们对于GCs与GR互相作用的更深入地研究以及不断涌现的新药物的实验进展,GR配体研究将进入新的发展阶段。
所有作者均声明不存在利益冲突


























