
幼年皮肌炎(JDM)是儿童时期最常见的特发性炎症性肌病,主要临床表现为特征性皮疹、四肢对称性近端肌无力和肌酶升高,也可累及肺部、消化道等重要脏器。肌炎特异性抗体已成为JDM的特异性生物标志物,不同的临床亚型具有不同的临床表现、疾病进程、治疗反应及远期预后。对JDM病情的评估及治疗应答情况的评价尤为重要。JDM的治疗多采用联合治疗方案,包括糖皮质激素、免疫抑制剂、生物制剂或JAK抑制剂等小分子靶向药。本病异质性较大,急性期如病情控制不佳易导致肌肉萎缩、肌腱挛缩和钙质沉着等远期后遗症。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
幼年皮肌炎(juvenile dermatomyositis, JDM)是一种免疫介导的,以皮肤、横纹肌、肺和胃肠道等部位的急性和慢性非化脓性炎症为特征的多系统受累的疾病。其主要特点为皮肤、肌肉和主要脏器的小血管炎症。典型临床表现包括眼睑紫红色皮疹、Gottron征、四肢对称性近端肌无力以及包括肌酸激酶(creatine kinase,CK)在内的肌酶显著升高[1]。不同的血清学和临床亚型具有不同的临床表现、疾病进程、治疗反应及远期预后。各种生物制剂及小分子靶向药物的出现为难治性JDM的治疗提供了更好的选择。为提高国内儿科及风湿科医师对不同亚型JDM的认识及诊断治疗水平,中华医学会儿科学分会风湿病学组等学术组织特制定了《幼年皮肌炎诊断与治疗专家共识》。
国外研究资料显示,JDM占所有幼年特发性炎症性肌病患儿的81.2%~85.0%,其发病率为每年1.9/100万~4.0/100万,患病率约为每年2.5/100万[2]。JDM的发病年龄和确诊年龄的中位数分别为5.7~6.9岁和7.4~7.7岁,但约1/4的患儿发病年龄<4岁[3]。JDM患儿男女比例为2.3∶1.0,男童较女童更易患本病[4]。目前国内尚未见完善的JDM流行病学调查数据。
目前JDM的病因尚未完全阐明,但与遗传因素和环境因素有关。全基因组分析提示在人类白细胞相关抗原区域、细胞因子基因和淋巴细胞信号基因中存在JDM的易感和保护性等位基因[5]。环境因素包括柯萨奇病毒、流感病毒、微小病毒、乙型肝炎病毒、A群链球菌、弓形虫和螺旋体感染以及疫苗、紫外线照射和药物等[6]。
巨噬细胞、T淋巴细胞、浆细胞样树突状细胞和自身抗体在JDM的发病中发挥一定作用。CD4+ T淋巴细胞是JDM血管周围和肌周组织中淋巴细胞浸润的主要细胞成分。B淋巴细胞和浆细胞在自身抗体产生中起主要作用。超过60%的JDM患儿存在自身抗体[6]。自身抗体分为肌炎特异性抗体和肌炎相关性抗体。每个JDM患儿通常仅有1种肌炎特异性抗体阳性,具有相同肌炎特异性抗体的儿童通常有相似的临床特征和长期预后[7]。近年来,研究发现Ⅰ型干扰素(IFN)在JDM的发病机制中发挥重要作用[8]。
JDM起病多缓慢,临床症状逐渐明显并趋于典型。本病通常表现为易疲劳、肌肉无力和皮疹,有时表现为发热、咳嗽、腹痛、吞咽困难、肌肉疼痛和关节炎等。部分病例全身症状重,病情进展迅速,合并呼吸衰竭和心功能不全而死亡。
皮疹可与肌无力同时出现,或发生在肌肉症状后数周,偶有以皮疹为首发症状的病例。典型的皮肤改变为上眼睑或上、下眼睑紫红色斑疹伴轻度水肿[9,10]。皮疹可逐渐蔓延及前额、鼻梁、上颌骨部位,内眦及眼睑部位可见毛细血管扩张。颈部和上胸部"V"字区、躯干部及四肢伸侧等处可出现弥漫性或局限性暗红色斑。部分皮疹消退后可留有色素沉着。
另一类特征性皮肤改变为Gottron征,此类皮疹见于掌指/跖趾关节和指/趾间关节伸面,亦可出现于肘、膝和踝关节伸侧。皮疹呈红色或紫红色,黄豆大小,部分可融合成块状,可伴细小鳞屑。随着时间进展局部出现皮肤萎缩及色素减退。约46%的患儿在甲襞可见僵直的毛细血管扩张,其上常见瘀点,这一改变也是JDM的特征性改变。甲襞变化是小血管炎症的证据,并且可能与皮肤和肌肉疾病活动有关,甲襞变化也与较低的儿童肌炎评估量表(childhood myositis assessment scale,CMAS)评分、较高的CK水平有关[11]。部分患儿可出现"技工手",表现为手指末端皮肤粗糙、皲裂。
严重和迁延不愈的JDM患儿常发生皮肤溃疡,这可能提示预后不良,眼角部、腋窝、肘部或受压部位出现血管炎性溃疡是本病严重的并发症,继发感染后治疗非常困难。溃疡在病理学上是皮肤血管病变的结果,由小血管缺氧和缺血引起,并且可能提示其他系统有类似的血管病变[3,12,13]。
少见的皮肤改变可有斑秃,这一改变并非本病特有,系统性红斑狼疮的患儿也可以出现。其他一些非特异性改变包括受累肢体的皮肤变薄和表皮变光滑,慢性病例可出现局部皮肤和皮下组织萎缩。
肌无力是JDM的主要特征,通常累及横纹肌,任何部位的肌肉均可受累,肢带肌、四肢近端及颈前屈肌最常受累,并以四肢近端和颈部肌肉受累更为严重,受累肌肉早期出现水肿和硬结,晚期可出现肌肉萎缩。临床表现主要为渐进性、对称性肌无力,也可伴有肌肉疼痛。病初患儿可表现为上楼困难、不能蹲下、穿衣困难等,进而发展为坐、立、行动和翻身困难,小年龄组患儿可仅表现为频繁跌倒。颈前屈肌无力表现为平卧时不能将颈部前屈,呈"后滴状征"阳性。涉及眼、舌、软腭时可致眼睑下垂、斜视、吞咽困难、呛咳等。肋间肌和膈肌、腹肌受累时,可引起呼吸困难而危及生命。晚期可因关节周围肌肉萎缩导致屈曲挛缩、活动受限、功能障碍。
肺部受累与预后不良有密切关系,可表现为发音困难、呼吸困难、肺功能检查(pulmonary function testing,PFT)异常、间质性肺病(interstitial lung disease,ILD)和气胸[14]。7%~19%的JDM患儿可出现ILD或快速进展性ILD而危及生命,尤其常发生在临床无肌病性皮肌炎(clinically amyopathic dermatomyositis,CADM)、血清抗黑色素瘤分化相关基因5(melanoma differentiation associated gene 5,MDA5)抗体阳性和抗合成酶抗体阳性的患儿中[15]。其中37%的患儿肺高分辨率CT发现异常,但大多无临床症状。其他影像学异常包括肺部结节、不规则磨玻璃影、纤维化和支气管壁增厚。
食管和胃肠是本病常见的受累器官,可因肌肉病变导致食道运动异常,出现肠壁肿胀,严重时合并溃疡、出血甚至穿孔。
心脏方面可见心脏增大、心电图异常,严重者可因心肌炎、心律失常、心功能不全而死亡。
钙质沉着是JDM严重的并发症之一,也是JDM的特殊表现。钙质沉着最早可发生于病程6个月内,也可发生于起病后10~20年。病理改变常发生于皮肤和皮下组织或较深层的筋膜和肌肉,表现为皮下小硬块或结节、关节附近团块状沉着、肌肉筋膜片状钙化等。这些变化可引起肢体酸痛、关节挛缩和功能障碍。钙化区常形成溃疡,并渗出白色石灰样物质。钙质沉着部位也可继发感染。广泛钙化最常发生于未治疗或未充分治疗而病程迁延和进展的患儿,但部分患儿虽已接受积极治疗,在疾病后期仍有可能发生钙质沉着。与钙质沉着相关的危险因素包括诊断延误、治疗延迟、疾病活动度高、发病年龄较小和心脏受累等。
中枢神经系统受累患儿头颅磁共振成像(MRI)检查可见脱髓鞘改变。眼部症状可出现视网膜绒毛状渗出、色素沉着、视乳头萎缩、水肿出血或视神经纤维变性。部分患儿还可并发脂肪代谢障碍,表现为局限性或广泛性皮下脂肪消失。脂肪营养不良发生在8%~14%的JDM中,可导致皮下和内脏脂肪总体、部分或局部进行性减少。肌肉萎缩、肌腱挛缩和由此造成的关节功能障碍是本病常见的远期并发症。
血清肌酶水平升高是肌肉损伤的标志,包括CK、乳酸脱氢酶、谷丙转氨酶和谷草转氨酶等,以CK为主。然而,非急性炎症期,肌酶水平正常,并不能排除JDM诊断。定期复查有助于了解病情的演变、疗效监测及预后评价。
幼年特发性炎症性肌病患儿血清中有多种肌炎特异性抗体(myositis-specific autoantibodies,MSA)和肌炎相关性抗体(myositis-asso-ciated autoantibodies,MAA)。常见的MSA出现在45%~55%的患儿中,MAA出现在16%~20%的患儿中[16]。MSA的发现提供了有关疾病亚型和预后的重要信息,目前已成为预测幼年特发性炎症性肌病临床表型及预后的重要生物标志物,见表1。
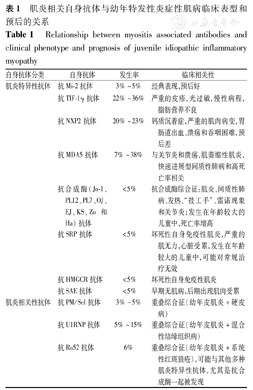
肌炎相关自身抗体与幼年特发性炎症性肌病临床表型和预后的关系
Relationship between myositis associated antibodies and clinical phenotype and prognosis of juvenile idiopathic inflammatory myopathy
肌炎相关自身抗体与幼年特发性炎症性肌病临床表型和预后的关系
Relationship between myositis associated antibodies and clinical phenotype and prognosis of juvenile idiopathic inflammatory myopathy
| 自身抗体分类 | 自身抗体 | 发生率 | 临床相关性 |
|---|---|---|---|
| 肌炎特异性抗体 | 抗Mi-2抗体 | 3%~5% | 经典表现,预后好 |
| 抗TIF-1γ抗体 | 22%~36% | 严重的皮疹,光过敏,慢性病程,脂肪营养不良 | |
| 抗NXP2抗体 | 20%~23% | 钙质沉着症,严重的肌肉病变,胃肠道出血、溃疡和吞咽困难,预后差 | |
| 抗MDA5抗体 | 7%~38% | 与关节炎和溃疡,肌萎缩性肌炎,快速进展型间质性肺病和高死亡率相关 | |
| 抗合成酶(Jo-1、PL12、PL7、OJ、EJ、KS、Zo和Ha)抗体 | <5% | 抗合成酶综合征:肌炎、间质性肺病、发热、"技工手"、雷诺现象和关节炎;发生在年龄较大的儿童中,死亡率增高 | |
| 抗SRP抗体 | <5% | 坏死性自身免疫性肌炎,严重的肌无力,心脏受累,发生在年龄较大的儿童中,可能对常规治疗无效 | |
| 抗HMGCR抗体 | <5% | 坏死性自身免疫性肌炎 | |
| 抗SAE抗体 | <5% | 早期无肌病,后期出现肌肉受累 | |
| 肌炎相关性抗体 | 抗PM/Scl抗体 | 3%~5% | 重叠综合征(幼年皮肌炎+硬皮病) |
| 抗U1RNP抗体 | 5%~15% | 重叠综合征(幼年皮肌炎+混合性结缔组织病) | |
| 抗Ro52抗体 | 6% | 重叠综合征(幼年皮肌炎+系统性红斑狼疮),可能与其他多种肌炎特异性抗体,尤其是抗合成酶一起被发现 |
红细胞沉降率在疾病进展期可出现升高,与疾病的活动性相关。Krebs von den Lungen-6(KL-6)是一种黏蛋白样糖蛋白,主要表达在Ⅱ型肺泡上皮细胞和支气管上皮细胞。抗MDA5抗体阳性皮肌炎(DM)/JDM患者的血清铁蛋白、KL-6和白细胞介素(IL)-18水平通常升高,而且与疾病活动性呈正相关。尤其是合并ILD的DM/JDM患者,经过治疗,血清铁蛋白、KL-6和IL-18水平可下降。因此,血清铁蛋白、KL-6和IL-18可作为监测疾病活动性及预测预后的重要指标。
典型的肌源性损害表现为插入电位增加、纤颤波、正锐波;收缩时呈短时限、低振幅、多相性电位;自发异常高频放电。
较少应用,但对不典型病例、治疗反应差的患儿具有重要的诊断价值。在肌肉受累最突出部位取材可能获得更高的阳性率,通常取股四头肌和三角肌等。JDM的特征性组织学表现包括束周萎缩、肌纤维变性和再生、血管周围不同程度炎性细胞浸润、内皮肿胀和坏死、主要组织相容性复合体(major histocompatibility complex,MHC)Ⅰ类分子过表达。
X线片可评估软组织和关节周围钙化的范围、程度。肺高分辨CT联合肺功能检查可发现患儿早期肺损害,主要表现为肺间质性改变,以小叶内间质增生和磨玻璃影最常见。肌肉MRI检查T2加权脂肪抑制序列成像可显示肌肉炎症呈水肿高信号表现,对早期肌肉病变敏感,而T1加权序列通常用于检测肌肉萎缩、肌内脂肪积聚或纤维化部分[17]。肌酶正常的患儿MRI可有阳性改变。全身MRI可以提供受影响肌肉分布模式的全面图像,并揭示临床上未预料到的远端或轴向肌肉群的受累情况,正成为一种确定肌病范围和随访JDM患儿的重要方法[18]。
可发现毛细血管袢扭曲、管壁增厚、周围血管缺失或毛细血管袢呈树枝状簇集等现象[19]。
目前临床工作中仍沿用1975年Bohan和Peter[9,10]制定的JDM分类方案和诊断标准:(1)典型的皮肤改变,包括上眼睑皮肤呈紫红色伴眼眶周围水肿(向阳征)以及掌指关节和近端指间关节背侧有红色鳞屑样皮疹(Gottron征);(2)对称性近端肌无力,可伴吞咽困难及呼吸肌无力;(3)实验室检查:血清骨骼肌酶活性升高,尤其是CK、谷草转氨酶;(4)肌电图异常:电位、短时限多相波;纤颤电位、阳性棘波、插入电位延长;安静时高波幅异常放电等;(5)肌肉活检异常:肌纤维变性、坏死,细胞吞噬、再生、嗜碱性变,核膜变大,核膜明显,筋膜周围结构萎缩,纤维大小不一,伴炎性渗出。
具备第(1)项及(2)~(5)项中的3项以上,可确诊为JDM,若缺乏第(1)项,具备(2)~(5)项中的3项以上,诊断为多发性肌炎。该诊断标准简单实用,但无法满足对疾病进行临床分型的需求。
欧洲抗风湿病联盟/美国风湿病学会(EULAR/ACR)于2017年制定了成人和儿童特发性炎症性肌病的分类标准[20]。该标准按照是否进行肌肉活检分别对皮疹、肌无力、其他临床表现及实验室检查作了不同权重计分(表2)。
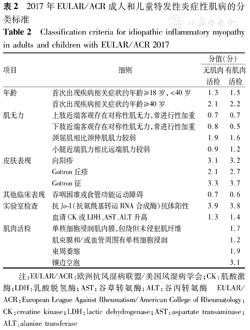
2017年EULAR/ACR成人和儿童特发性炎症性肌病的分类标准
Classification criteria for idiopathic inflammatory myopathy in adults and children with EULAR/ACR 2017
2017年EULAR/ACR成人和儿童特发性炎症性肌病的分类标准
Classification criteria for idiopathic inflammatory myopathy in adults and children with EULAR/ACR 2017
| 项目 | 细则 | 分值(分) | |
|---|---|---|---|
| 无肌肉活检 | 有肌肉活检 | ||
| 年龄 | 首次出现疾病相关症状的年龄≥18岁,<40岁 | 1.3 | 1.5 |
| 首次出现疾病相关症状的年龄≥40岁 | 2.1 | 2.2 | |
| 肌无力 | 上肢近端客观存在对称性肌无力,常进行性加重 | 0.7 | 0.7 |
| 下肢近端客观存在对称性肌无力,常进行性加重 | 0.8 | 0.5 | |
| 颈屈肌相比颈伸肌肌力较弱 | 1.9 | 1.6 | |
| 小腿近端肌力相比远端肌力较弱 | 0.9 | 1.2 | |
| 皮肤表现 | 向阳疹 | 3.1 | 3.2 |
| Gottron丘疹 | 2.1 | 2.7 | |
| Gottron征 | 3.3 | 3.7 | |
| 其他临床表现 | 吞咽困难或食管功能运动障碍 | 0.7 | 0.6 |
| 实验室检查 | 抗Jo-1(抗氨酰基转运RNA合成酶)抗体阳性 | 3.9 | 3.8 |
| 血清CK或LDH、AST、ALT升高 | 1.3 | 1.4 | |
| 肌肉活检 | 单核细胞浸润肌内膜,包绕但未侵犯肌纤维 | 1.7 | |
| 肌束膜和/或血管周围有单核细胞浸润 | 1.2 | ||
| 束周萎缩 | 1.9 | ||
| 镶边空泡 | 3.1 | ||
注:EULAR/ACR:欧洲抗风湿病联盟/美国风湿病学会;CK:肌酸激酶;LDH:乳酸脱氢酶;AST:谷草转氨酶;ALT:谷丙转氨酶
EULAR/ACR:European League Against Rheumatism/American College of Rheumatology;CK:creatine kinase;LDH:lactic dehydrogenase;AST:aspartate transaminase;ALT:alanine transferase
具体分类诊断标准为:(1)确诊特发性炎症性肌病(可能性≥90%):无肌肉活检者评分≥7.5分,有肌肉活检者评分≥8.7分;(2)很可能为特发性炎症性肌病(55%≤可能性<90%):无肌肉活检者评分≥5.5分,有肌肉活检者评分≥6.7分。符合上述分类诊断标准,如果起病年龄<18岁,存在向阳疹(上眼睑紫红色皮疹伴眶周水肿)、Gottron丘疹(指关节背面红斑状鳞屑性丘疹)或Gottron征(指关节背面的扁平红斑疹)时,诊断为JDM;如果患儿没有上述任何皮肤表现,则诊断为幼年多发性肌炎。
JDM需与以下几种疾病进行鉴别诊断。
病毒感染,如流感、柯萨奇病毒感染可出现急性短暂性肌炎,可有一过性血清CK增高,常于3~5 d恢复。近期感染史和短期自限性病程有助于与本病鉴别。
应与无皮疹的多发性肌炎鉴别。本病的特征为全身广泛性肌无力,受累肌肉在持久或重复活动后肌无力加重,多伴有眼睑下垂,有晨轻暮重的特点。可行抗乙酰胆碱受体抗体测定和新斯的明试验进行鉴别。
应与无皮疹的多发性肌炎鉴别。患儿往往存在其他家庭成员受累的家族史,男性发病,有典型的鸭步及腓肠肌假性肥大,可通过基因检测确诊。
是一组由于糖、脂肪、线粒体代谢异常引起组织细胞产能障碍,造成以反复肌无力、运动不耐受为主要临床表现的肌肉疾病。这些疾病的诊断需经肌肉活检免疫组织化学、酶学检测及基因检测确诊。
若JDM以关节炎为主要表现,易与幼年特发性关节炎相混淆。系统性红斑狼疮、硬皮病、混合性结缔组织病等亦可出现肌炎表现,但每种疾病均有其特征性的临床表现,故这些疾病与JDM之间的鉴别通常并不困难,但也可能发生重叠综合征。抗MDA5抗体阳性的JDM易出现皮肤溃疡和网状青斑等临床表现,应注意及时完善基因检测与自身炎症性疾病中的IFN通路病进行鉴别诊断。
由于JDM临床表现多种多样,全身重要脏器受累程度不等,因此对JDM患儿病情进行准确的评估非常必要。
目前国际上针对JDM患儿整体状况、肌肉和皮肤受累的严重程度、生活质量等采用一系列评分方法进行综合评估[21,22,23,24]。
医师/家长/患儿整体评估采用10 cm视觉模拟评分(visual analog scale,VAS);活动性整体评估采用疾病活动性评分(disease activity score,DAS)或肌炎活动性评估(myositis disease activity assessment,MDAA);损害整体评估采用肌炎损害指数(myositis damage index,MDI)[25]。
8组肌群的标准化对抗型徒手肌力检查(manual muscle testing, MMT)评分(MMT8)已被验证可用于评估JDM患儿的肌力[26]。CMAS是一种客观测量肌力的临床评估工具。通过让儿童完成一些不同的躯体动作(主要评估中轴肌群和近端肌群)进行CMAS评分,从而检测肌肉的功能和耐力[27]。评分为0~52分,评分越高表明肌力越强。但部分CMAS的动作完成还取决于年龄,低年龄的患儿评估出来的分数可能比实际分数要低,因此临床应用时需结合其他评估方法。
皮肤评估工具(cutaneous assessment tool,CAT)、皮肤DAS、皮肌炎皮肤严重指数等多种工具和方法可用于JDM患儿皮肤活动性的评估。甲襞毛细血管镜是最常用的CAT,用于检测指甲周围毛细血管变化,甲襞毛细血管密度是一个敏感的测量皮肤和肌肉疾病活动状态的指标[28]。
采用儿童健康评价问卷(childhood health assessment questionnaire,CHAQ)和儿童健康问卷(child health questionnaire,CHQ)等进行功能评估。
多脏器损害是JDM的重要临床特征,JDM相关的ILD、心脏病、消化道病变较为常见。因此,在诊断及随诊过程中务必行肺高分辨率CT、心电图、心脏彩超和消化道B超等检查来评估脏器受累的情况,合并肺受累的患儿还需行肺功能检查,合并消化道受累的患儿需酌情行消化道造影。钙化是JDM患儿严重的并发症,随诊过程中应通过仔细的查体或影像学检查及时发现钙化。
2017年欧洲儿童风湿病单中心接入点(single hub and access point for pediatric rheumatology in Europe,SHARE)发表的JDM管理专家共识中提出了影响重症JDM患儿的高危病征,具体包括:(1)严重的肌无力(因肌无力卧床);(2)CMAS评分<15分或MMT8<30分;(3)具有呼吸肌无力或吞咽困难;(4)消化系统血管炎(存在影像学证据或出现便血);(5)心肌炎;(6)肺受累;(7)中枢神经系统疾病(意识障碍或惊厥);(8)皮肤溃疡;(9)需要进行重症监护;(10)年龄<1岁。具有高危病征的JDM患儿应早期积极治疗。
2016年国际肌炎评估及临床研究小组(the international myositis assessment and clinical studies group,IMACS)与国际儿童风湿病试验组织(the paediatric rheumatology international trials organisation,PRINTO)共同制定了JDM的临床应答的标准,见表3[29]。总改善分数是每个核心集改善分数的总和。总改善分数≥30分代表最小的改善,≥45分代表中度改善,≥70分代表最大改善。
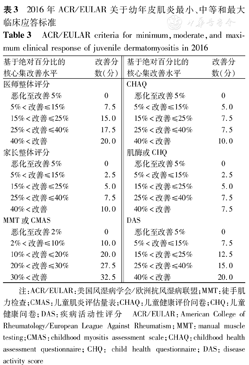
2016年ACR/EULAR关于幼年皮肌炎最小、中等和最大临床应答标准
ACR/EULAR criteria for minimum,moderate,and maximum clinical response of juvenile dermatomyositis in 2016
2016年ACR/EULAR关于幼年皮肌炎最小、中等和最大临床应答标准
ACR/EULAR criteria for minimum,moderate,and maximum clinical response of juvenile dermatomyositis in 2016
| 基于绝对百分比的核心集改善水平 | 改善分数(分) | 基于绝对百分比的核心集改善水平 | 改善分数(分) | ||
|---|---|---|---|---|---|
| 医师整体评分 | CHAQ | ||||
| 恶化至改善5% | 0 | 恶化至改善5% | 0 | ||
| 5%<改善≤15% | 7.5 | 5%<改善≤15% | 5.0 | ||
| 15%<改善≤25% | 15.0 | 15%<改善≤25% | 7.5 | ||
| 25%<改善≤40% | 17.5 | 25%<改善≤40% | 7.5 | ||
| 40%<改善 | 20.0 | 40%<改善 | 10.0 | ||
| 家长整体评分 | 肌酶或CHQ | ||||
| 恶化至改善5% | 0 | 恶化至改善5% | 0 | ||
| 5%<改善≤15% | 2.5 | 5%<改善≤15% | 2.5 | ||
| 15%<改善≤25% | 5.0 | 15%<改善≤25% | 5.0 | ||
| 25%<改善≤40% | 7.5 | 25%<改善≤40% | 7.5 | ||
| 40%<改善 | 10.0 | 40%<改善 | 7.5 | ||
| MMT或CMAS | DAS | ||||
| 恶化至改善2% | 0 | 恶化至改善5% | 0 | ||
| 2%<改善≤10% | 10.0 | 5%<改善≤15% | 7.5 | ||
| 10%<改善≤20% | 20.0 | 15%<改善≤25% | 12.5 | ||
| 20%<改善≤30% | 27.5 | 25%<改善≤40% | 15.0 | ||
| 30%<改善 | 32.5 | 40%<改善 | 20.0 | ||
注:ACR/EULAR:美国风湿病学会/欧洲抗风湿病联盟;MMT:徒手肌力检查;CMAS:儿童肌炎评估量表;CHAQ:儿童健康评价问卷;CHQ:儿童健康问卷;DAS:疾病活动性评分
ACR/EULAR:American College of Rheumatology/European League Against Rheumatism;MMT:manual muscle testing;CMAS:childhood myositis assessment scale;CHAQ:childhood health assessment questionnaire;CHQ: child health questionnaire;DAS:disease activity score
有吞咽困难者及时予鼻饲防止误吸;避免紫外线暴露;注意保护患儿,防止因肌力减弱、活动不便造成的外伤;合并钙化及皮肤溃疡的患儿注意预防感染;急性期过后应尽早进行合理的康复锻炼,避免肌肉萎缩、肌腱和关节挛缩。
主要采用糖皮质激素(简称"激素")联合免疫抑制剂的治疗方案:初始治疗使用泼尼松/甲泼尼龙和甲氨蝶呤(Methotrexate,MTX)等;对于重症或存在高危病征的患儿以及难治性、对MTX反应不佳、初始治疗疗效不好的低龄患儿或有不良反应者可采用激素联合丙种球蛋白、环孢素A(Cyclosporine A, CsA)或硫唑嘌呤(Azathioprine,AZA)、环磷酰胺(Cyclophosphamide,CTX)、霉酚酸酯(Mycophenolate mofetil,MMF)等药物治疗,病情仍难控制者可联合应用沙利度胺、生物制剂或JAK抑制剂等。
激素目前仍是治疗JDM的首选药物。一般初始剂量为泼尼松1~2 mg/(kg·d),最大剂量60 mg/d。病情进展迅速或有呼吸困难、吞咽困难、心肌损伤及消化道血管炎者,可采用大剂量甲泼尼龙静脉冲击治疗,剂量为10~30 mg/(kg·d)(最大剂量1 g/d),连用3~5 d,随后口服泼尼松治疗,如果病情控制不佳,隔3~5 d后可再予3 d甲泼尼龙静脉冲击治疗。
口服应用足量激素4周后逐渐开始减量,具体减量过程视病情缓解情况而定,通常激素总疗程1~2年,部分病情反复的患儿激素应用时间长达数年。在治疗过程中应注意长期使用激素的不良反应,如感染、骨质疏松、白内障和生长发育迟缓等。
激素与免疫抑制剂的联用可提高疗效,减少激素用量,避免不良反应。
MTX对控制肌肉的炎症和改善皮肤症状均有帮助,是免疫抑制剂中的首选药物。多采用口服给药,剂量10~15 mg/m2,每周应用1次,最大量为15 mg。主要不良反应为肝功能受损、骨髓抑制、口腔炎等。用药期间可同步服用叶酸避免口腔炎发生,应定期监测肝肾功能和血常规。
主要用于激素或MTX治疗无效的难治病例,肺间质病变也是用药的适应证。常用剂量为2~3 mg/(kg·d),最大剂量为100 mg/d,分2~3次口服。主要不良反应为高血压、多毛、胃肠道症状、齿龈增生及肾脏毒性等。长期用药的患儿需要监测药物的谷浓度以防止药物中毒。
MMF常用剂量为30~50 mg/(kg·d),分2次或3次口服,最大量为1.5 g/d。常见不良反应为胃肠道反应和血细胞减少等。对激素及MTX治疗效果欠佳的JDM患儿推荐应用吗替麦考酚酯治疗。
多采用静脉冲击疗法,主要用于肺间质病变或中枢神经系统受累的患儿,剂量为200~400 mg/(m2·次),每4周用药1次,单次最大剂量为600 mg,根据病情缓解程度调整用药的间隔时间。主要不良反应为骨髓抑制、出血性膀胱炎、性腺抑制、血细胞减少等。用药期间应注意给予充分的水化和碱化,需监测血常规和肝肾功能。
AZA用于MTX或CsA治疗无效者,常用剂量为1~3 mg/(kg·d),分2~3次口服,最大量为100 mg/d。主要不良反应为骨髓抑制、血细胞减少、转氨酶升高等。用药时应定期复查血常规和肝肾功能等。
适用于起病时较重或疾病进展迅速的患儿、激素无效或同时联合免疫抑制剂治疗效果欠佳者。剂量为400 mg/(kg·d),最大剂量为15 g/d,可连用3~5 d,必要时每月应用1次,连续应用3~6个月或更长时间对肌力和皮疹均有明显改善效果。
HCQ属于抗疟药,适用于皮肤病变明显者,剂量为5.0~6.5 mg/(kg·d),可顿服或分2次服用,最大量为0.3 g/d。不良反应主要为视野缺损、粒细胞减少、肝功能受损等,>6岁且能配合行视野检查的患儿可考虑应用,应用过程中应定期监测视野。
沙利度胺具有特异性免疫调节作用,能抑制单核细胞产生肿瘤坏死因子(TNF),还能协同刺激T淋巴细胞、辅助T淋巴细胞应答,并可抑制血管形成和黏附分子的活性。沙利度胺对难治性JDM及合并钙化的患儿有明显的效果。一般3岁以上儿童考虑应用,1~2 mg/(kg·d),最大剂量为25 mg/次,3次/d口服。主要不良反应为末梢神经炎、便秘和嗜睡,因其可造成"海豚儿",禁用于怀孕的患者。
近年来,生物制剂开始用于治疗重症、难治性JDM,一些个案报道使用抗CD20单克隆抗体、抗CTAL-4单克隆抗体、TNF-α抑制剂、IL-6单克隆抗体有一定疗效,但目前尚未见大样本随机对照研究来进一步证实。
JAK抑制剂可减少IFN诱导的STAT1磷酸化,并阻断JAK-STAT通路。Sabbagh等[30]的研究表明应用JAK抑制剂治疗2例抗MDA5抗体阳性、血液IFN反应基因特征升高以及合并ILD的难治性JDM患儿,治疗后病情得到极大改善,显著减少了糖皮质激素及CTX、MMF的使用。国内单中心研究表明JAK抑制剂对于JDM的肌无力症状和皮疹均有明显改善作用[31]。托法替布可参考治疗幼年特发性关节炎的剂量[32](表4),每次最大剂量为5 mg,每日2次口服。巴瑞替尼剂量为0.04 mg/kg,最大剂量为2 mg/d。芦可替尼剂量为:体质量<25 kg,5 mg/d;体质量≥25 kg,10 mg/d。治疗过程中需注意监测血常规,注意预防感染。
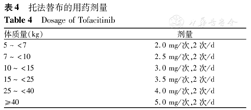
托法替布的用药剂量
Dosage of Tofacitinib
托法替布的用药剂量
Dosage of Tofacitinib
| 体质量(kg) | 剂量 |
|---|---|
| 5~<7 | 2.0 mg/次,2次/d |
| 7~<10 | 2.5 mg/次,2次/d |
| 10~<15 | 3.0 mg/次,2次/d |
| 15~<25 | 3.5 mg/次,2次/d |
| 25~<40 | 4.0 mg/次,2次/d |
| ≥40 | 5.0 mg/次,2次/d |
治疗JDM皮下钙化患儿除抗炎治疗外,可采用钙通道阻滞剂、二膦酸盐、硫代硫酸钠、氢氧化铝和丙磺舒等药物干预钙磷代谢,以达到减少钙质沉积、溶解已沉积钙质的目的。
皮肤病变严重者可局部外用药物治疗。严重的钙质沉着可影响病灶局部关节或脏器功能,可考虑外科手术治疗。皮肤溃疡合并感染者需积极抗感染治疗。
JDM是一种异质性非常大的疾病,部分患儿疾病活动期为2年,经过治疗可得到完全缓解,部分患儿可有多次复发或呈慢性持续状态,病情可持续3~5年或更久。本病最常见的死亡原因为肺部感染、胃肠道出血及穿孔。最常见的后遗症为因急性期病情控制不佳导致的肌肉萎缩、肌腱挛缩和钙质沉着。
(张俊梅 邓江红 檀晓华 李彩凤 执笔)
参与本共识讨论、制定的专家(按姓氏拼音排序):曹兰芳(上海交通大学医学院附属仁济医院);陈同辛(上海交通大学医学院附属上海儿童医学中心);陈雨青(安徽省儿童医院);邓江红(国家儿童医学中心,首都医科大学附属北京儿童医院);封其华(苏州大学附属儿童医院);韩梅(大连市儿童医院);金燕樑(上海交通大学医学院附属上海儿童医学中心);李彩凤(国家儿童医学中心,首都医科大学附属北京儿童医院);李亚蕊(山西省儿童医院);李玉峰(上海交通大学医学院附属新华医院);李志辉(湖南省儿童医院);廖亚彬(昆明市儿童医院);刘小惠(江西省儿童医院);刘翠华(郑州大学附属儿童医院,河南省儿童医院,郑州儿童医院);卢美萍(浙江大学医学院附属儿童医院);孙利(复旦大学附属儿科医院);檀晓华(国家儿童医学中心,首都医科大学附属北京儿童医院);尹薇(武汉妇女儿童医院);张洪霞(山东大学齐鲁儿童医院);张俊梅(国家儿童医学中心,首都医科大学附属北京儿童医院);郑雯洁(温州医科大学附属第二医院);周志轩(首都儿科研究所附属儿童医院)
所有作者均声明不存在利益冲突

























