
探究病因不明的早发癫痫性脑病(EOEE)的遗传学病因及其在早期诊断中的价值。
前瞻性收集2018年1月至2021年1月就诊于福建省立医院门诊及住院部的60例病因不明的EOEE患儿的资料,采集外周血进行全外显子组测序及拷贝数变异(CNV)检测,分析患儿临床特点、遗传学测序结果。
检测出EOEE相关的致病或可疑致病突变24例,包括婴儿痉挛症(10例)、Dravet综合征(3例)、吡哆醇依赖性癫痫及大田原综合征(各1例)、非已知癫痫性脑病(9例)。患儿起病年龄1 d~11个月(中位年龄4.2个月),就诊年龄2 d~4岁(中位年龄10个月),确诊年龄均控制在就诊1个月以内。发现单基因变异20例(33.3%),CNV 4例(6.7%);涉及13种基因:KCNQ2、SCN1A、SCN8A、CACNA1E、CDKL5、PPP3CA、PCDH19、TSC1、TSC2、ZEB2、ALDH7A1、DCX、HNRNPU;4例CNV异常分别为17p13.3缺失、11q23.3q25缺失、1q36.31-p36.33缺失、1q43-1q44缺失合并Xp22.33重复;共20种变异为国内外首次报道的新发位点;11q23.3q25缺失导致婴儿痉挛症为国内外首次报道;ZEB2变异导致婴儿痉挛症、PPP3CA基因导致癫痫性脑病均系国内首例报道。
基因与CNV是EOEE患儿的重要潜在病因,病因不明时联合采用全外显子组测序和CNV测序技术检测,可提高EOEE患儿的遗传学病因诊断水平;对此类患儿早期进行遗传学检测,可以早期确诊及进行癫痫精准治疗。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
早发癫痫性脑病(early onset epilepticencephalopathy,EOEE)指一组在婴儿期起病的难治性癫痫,由于频繁的癫痫发作或癫痫放电导致严重认知和行为障碍、精神运动发育迟缓;包括早期肌阵挛脑病、大田原综合征、婴儿癫痫伴游走性局灶性发作、婴儿痉挛症(IS)、Dravet综合征、非已知的癫痫性脑病等[1];EOEE是预后最差的癫痫性脑病,基因突变是EOEE的重要病因[1]。基于此,本研究结合全外显子和全基因组拷贝数变异测序(copy number variations sequencing,CNV-seq)技术对60例不明原因的EOEE患儿进行基因变异检测,以期早期确诊及进行癫痫精准治疗。
前瞻性收集2018年1月至2021年1月就诊于福建省立医院门诊及住院部的60例病因不明的EOEE患儿详细资料,包括年龄、性别、起病年龄、家族史、围生史、生长发育史、发作形式、有无畸形、颅脑磁共振成像(MRI)、动态或视频脑电图、基因检测结果、血氨、乳酸、生化、血尿串联质谱筛查、治疗经过等。发育评估采用婴幼儿神经心理发育诊断量表,以末次评估为准;门诊或电话随访,至少随访6个月以上。本研究通过医院医学伦理委员会批准(批准文号:K201901015),患儿监护人均签署知情同意书。
(1)符合国际抗癫痫联盟(ILAE)制定的癫痫性脑病标准[2];(2)出生12个月内癫痫发作;(3)有多种癫痫发作类型;(4)常规抗癫痫药物难以控制发作;(5)认知功能、运动功能发育迟滞或倒退;(6)病因不明。除外代谢性疾病、围生期脑损伤、颅内感染等已知病因的患儿。
全外显子测序方法:取患儿外周静脉血2 mL,乙二胺四乙酸(EDTA)抗凝,提取DNA,样品检测及打断,构建文库,目的基因的捕获及富集,生物信息学分析,检索人类基因突变数据库(HGMD)、PubMed、Clinvar等数据库,参考《美国医学遗传学和基因组学学会变异分类指南》[3]对变异进行分类。CNV-seq检测方法:采集患儿外周静脉血2 mL,EDTA抗凝,提取DNA,测定浓度,构建DNA文库,应用NEXTSEQ 50测序仪上机测序,数据分析与计算,得出显著的拷贝数变异(CNV),在人类表型本体论(HPO)数据库中搜索以匹配相似的表型,同时进行定量聚合酶链反应(qPCR)实验排除二代测序的假阳性。最后采用Sanger测序验证父母及来源分析,以上检测均委托武汉康圣达医学检测所完成。
60例EOEE患儿基因诊断明确24例(40.0%),其中男、女各12例,起病年龄1 d~11个月(中位年龄4.2个月);就诊年龄2 d~4岁(中位年龄10个月)。IS 10例,Dravet综合征3例,吡哆醇依赖性癫痫1例,大田原综合征1例,非已知癫痫性脑病9例。6例有热性惊厥史(例3、6、12、14、18、23);家族史:热性惊厥史2例(例5大伯及祖父、例14父亲),癫痫病史4例(例18姐姐、例19外祖母、例13母亲、例22兄长)。24例患儿母孕史均未见明显异常。发作形式多样,以全面性强直阵挛、痉挛发作多见。治疗药物均为2种或以上,其中大部分未控制,仅例24发作控制。24例患儿在发病前后均存在不同程度的智力运动发育落后。
MRI正常11例,异常13例,分别为左右侧脑室内多发实体占位、皮质下层状异位、先天性无脑回畸形、胼胝体缺如各1例,室管膜下囊肿1例、脑积水8例。脑电图显示暴发抑制1例、典型或不典型高度失律伴或不伴多灶性棘(尖)慢波10例、多灶性棘(尖)慢波13例。
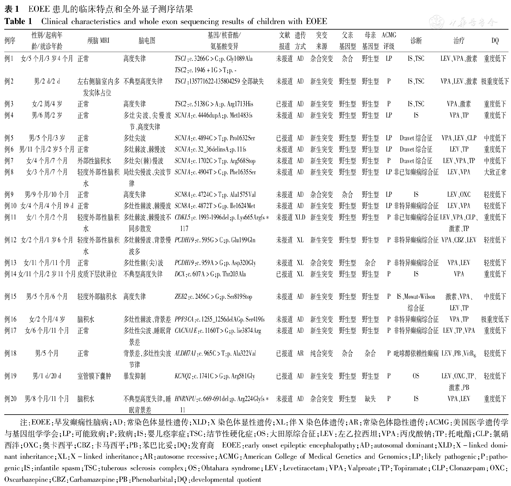
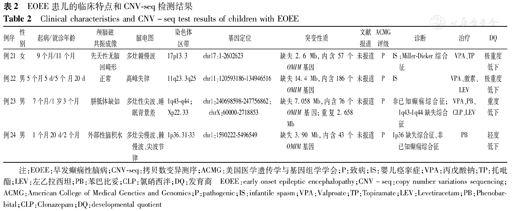
共检测出24种致病或可能致病基因变异,涉及13种基因,包括SCN1A、SCN8A、CACNA1E、CDKL5、PPP3CA、PCDH19、TSC1、TSC2、ZEB2、ALDH7A1、DCX、KCNQ2、HNRNPU,其中错义突变14例,移码突变4例,杂合缺失1例;15例为新生突变,5例为遗传性突变;15种变异位点既往国内外未见报道。其中单基因变异20例(33.3%,20/60例),4例(6.7%,4/60例)患儿共检出5种CNV异常,例23同时存在2种CNV变异,均为致病性变异。共20种为未报道的新发位点,11q23.3q25缺失导致IS为国内外首次报道,ZEB2变异导致IS、PPP3CA基因导致癫痫性脑病均系国内首例报道。患儿基因检测结果见表1、表2。
EOEE患儿的临床特点和全外显子测序结果
Clinical characteristics and whole exon sequencing results of children with EOEE
EOEE患儿的临床特点和全外显子测序结果
Clinical characteristics and whole exon sequencing results of children with EOEE
| 例序 | 性别/起病年龄/就诊年龄 | 颅脑MRI | 脑电图 | 基因/核苷酸/氨基酸变异 | 文献报道 | 遗传方式 | 突变来源 | 父亲基因型 | 母亲基因型 | ACMG评级 | 诊断 | 治疗 | DQ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 例1 | 女/5个月/3岁4个月 | 正常 | 高度失律 | TSC1:c.3266G>C;p.Gly1089Ala TSC2:c.1946+1G>T;p.- | 未报道 | AD | 杂合突变 | 杂合 | 野生型 | LP | IS、TSC | LEV、VPA、激素 | 重度低下 |
| 例2 | 男/2 d/2 d | 左右侧脑室内多发实体占位 | 不典型高度失律 | TSC1:135771622-135804259全部缺失 | 未报道 | AD | 新生突变 | 野生型 | 野生型 | P | IS、TSC | VPA、LEV、激素 | 极重度低下 |
| 例3 | 女/2周/4岁 | 正常 | 高度失律 | TSC2:c.5138G>A;p.Arg1713His | 已报道 | AD | 新生突变 | 野生型 | 野生型 | P | IS、TSC | VPA、激素 | 重度低下 |
| 例4 | 男/6周/2岁 | 正常 | 多灶尖波、尖慢波节、高度失律 | SCN1A:c.4446dupA;p.Met1483fs | 未报道 | AD | 新生突变 | 野生型 | 野生型 | LP | IS | VPA、TP | 重度低下 |
| 例5 | 男/5个月/3岁 | 正常 | 多灶尖波 | SCN1A:c.4894C>T;p.Pro1632Ser | 已报道 | AD | 新生突变 | 野生型 | 野生型 | LP | Dravet综合征 | VPA、LEV、CLP | 中度低下 |
| 例6 | 男/11个月/2岁5个月 | 正常 | 多灶棘波、棘慢波 | SCN1A:c.32_36delinsA;p.11fs | 未报道 | AD | 新生突变 | 野生型 | 野生型 | LP | Dravet综合征 | LEV、TP | 重度低下 |
| 例7 | 女/4个月/7个月 | 外部性脑积水 | 多灶尖(棘)慢波 | SCN1A:c.1702C>T;p.Arg568Stop | 未报道 | AD | 新生突变 | 野生型 | 野生型 | P | Dravet综合征 | LEV、VPA、TP | 中度低下 |
| 例8 | 女/3个月/7个月 | 轻度外部性脑积水 | 局灶尖慢波、尖波节律 | SCN1A:c.4904T>C;p.Phe1635Ser | 未报道 | AD | 新生突变 | 野生型 | 野生型 | LP | 非已知癫痫综合征 | LEV、VPA | 大致正常 |
| 例9 | 男/9个月/10个月 | 正常 | 高度失律 | SCN8A:c.4724C>T;p.Ala1575Val | 未报道 | AD | 杂合突变 | 杂合 | 野生型 | LP | IS | LEV、OXC | 轻度低下 |
| 例10 | 女/4个月/4个月19 d | 正常 | 多灶性棘波、棘慢波 | SCN8A:c.4872T>G;p.Ile1624Met | 未报道 | AD | 新生突变 | 野生型 | 野生型 | LP | 非特异癫痫综合征 | LEV、VPA | 轻度低下 |
| 例11 | 女/1个月/2个月 | 轻度外部性脑积水 | 多灶棘波、棘慢波不同步散发 | CDKL5:c.1993-1996del;p.Lys665Argfs*117 | 未报道 | XLD | 新生突变 | 野生型 | 野生型 | P | 非已知癫痫综合征 | LEV、VPA、CLP、激素、TP | 重度低下 |
| 例12 | 女/2个月/1岁6个月 | 轻度外部性脑积水 | 多灶棘慢波、背景慢波多 | PCDH19:c.595G>C;p.Glu199Gln | 未报道 | XL | 新生突变 | 野生型 | 野生型 | P | 非特异癫痫综合征 | VPA、CBZ、LEV | 轻度低下 |
| 例13 | 女/11个月/11个月 | 正常 | 多灶性棘(尖)波 | PCDH19:c.959A>G;p.Asp320Gly | 未报道 | XL | 杂合突变 | 野生型 | 杂合 | P | 非特异癫痫综合征 | VPA、LEV | 轻度低下 |
| 例14 | 女/11个月/2岁11个月 | 皮质下层状异位 | 不典型高度失律 | DCX:c.607A>G;p.Thr203Ala | 已报道 | XL | 新生突变 | 野生型 | 野生型 | P | IS | VPA | 重度低下 |
| 例15 | 男/5个月/6个月 | 轻度外部脑积水 | 高度失律 | ZEB2:c.2456C>G;p.Ser819Stop | 未报道 | AD | 新生突变 | 野生型 | 野生型 | P | IS、Mowat-Wilson综合征 | 激素、VPA、LEV、TP | 中度低下 |
| 例16 | 女/2个月/4岁 | 脑积水 | 多灶性棘波、背景差 | PPP3CA:c.1255_1256delAGp.Ser419fs | 未报道 | AD | 新生突变 | 野生型 | 野生型 | P | 非特异癫痫综合征 | VPA、TP | 极重度低下 |
| 例17 | 女/6个月/11个月 | 正常 | 多灶性尖波、睡眠背景差 | CACNA1E:c.1160T>G;p.lie3874Arg | 未报道 | AD | 新生突变 | 野生型 | 野生型 | P | 非特异癫痫综合征 | LEV、TP、VPA | 重度低下 |
| 例18 | 男/5个月 | 正常 | 背景差,多灶性尖波节律 | ALDH7A1:c.965C>T;p.Ala322Val | 已报道 | AR | 纯合突变 | 杂合 | 杂合 | P | 吡哆醇依赖性癫痫 | LEV、PB、VitB6 | 轻度低下 |
| 例19 | 男/1 d/20 d | 室管膜下囊肿 | 暴发抑制 | KCNQ2:c.1741C>G;p.Arg581Gly | 已报道 | AD | 新生突变 | 野生型 | 野生型 | P | OS | LEV、OXC、TP、激素、PB | 轻度低下 |
| 例20 | 男/8个月/11个月 | 脑积水 | 不典型高度失律,睡眠背景差 | HNRNPU:c.669-691del;p.Arg224Glyfs*11 | 未报道 | AD | 杂合突变 | 野生型 | 缺失 | P | IS | VPA、LEV | 重度低下 |
注:EOEE:早发癫痫性脑病;AD:常染色体显性遗传;XLD:X染色体显性遗传;XL:伴X染色体遗传;AR:常染色体隐性遗传;ACMG:美国医学遗传学与基因组学学会;LP:可能致病;P:致病;IS:婴儿痉挛症;TSC:结节性硬化症;OS:大田原综合征;LEV:左乙拉西坦;VPA:丙戊酸钠;TP:托吡酯;CLP:氯硝西泮;OXC:奥卡西平;CBZ:卡马西平;PB:苯巴比妥;DQ:发育商 EOEE:early onset epileptic encephalopathy;AD:autosomal dominant;XLD:X-linked dominant inheritance;XL:X-linked inheritance;AR:autosome recessive;ACMG:American College of Medical Genetics and Genomics;LP:likely pathogenic;P:pathogenic;IS:infantile spasm;TSC:tuberous sclerosis complex;OS:Ohtahara syndrome;LEV:Levetiracetam;VPA:Valproate;TP:Topiramate;CLP:Clonazepam;OXC:Oxcarbazepine;CBZ:Carbamazepine;PB:Phenobarbital;DQ:developmental quotient
EOEE患儿的临床特点和CNV-seq检测结果
Clinical characteristics and CNV-seq test results of children with EOEE
EOEE患儿的临床特点和CNV-seq检测结果
Clinical characteristics and CNV-seq test results of children with EOEE
| 例序 | 性别 | 起病/就诊年龄 | 颅脑磁共振成像 | 脑电图 | 染色体区带 | 基因定位 | 突变性质 | 文献报道 | ACMG评级 | 诊断 | 治疗 | DQ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 例21 | 女 | 9个月/11个月 | 先天性无脑回畸形 | 多灶棘慢波 | 17p13.3 | chr17:1-2602623 | 缺失2.6 Mb,内含57个OMIM基因 | 未报道 | P | IS;Miller-Dieker综合征 | VPA、TP | 极重度低下 |
| 例22 | 男 | 5个月5 d/5个月20 d | 正常 | 高峰失律 | 11q23.3q25 | chr11:120593186-134946516 | 缺失14.4 Mb,内含186个OMIM基因 | 未报道 | P | IS | VPA、激素、LEV | 极重度低下 |
| 例23 | 男 | 7个月/1岁3个月 | 胼胝体缺如 | 多灶性尖波、睡眠背景差 | 1q43-q44;Xp22.33 | chr1:240698598-247756862;chrX:60000-2718853 | 缺失7.058 Mb,内含76个OMIM基因;重复2.658 Mb | 未报道 | P | 非已知癫痫综合征;1q43-1q44缺失综合征 | VPA、PB、CLP、LEV | 重度低下 |
| 例24 | 男 | 1个月20 d/2个月 | 外部性脑积水 | 多灶尖慢波、棘慢波、尖波节律 | 1p36.31-33 | chr1:1590222-5496549 | 缺失3.90 Mb,内含43个OMIM基因 | 未报道 | P | 1p36缺失综合征、非已知癫痫综合征 | PB | 轻度低下 |
注:EOEE:早发癫痫性脑病;CNV-seq:拷贝数变异测序;ACMG:美国医学遗传学与基因组学学会;P:致病;IS:婴儿痉挛症;VPA:丙戊酸钠;TP:托吡酯;LEV:左乙拉西坦;PB:苯巴比妥;CLP:氯硝西泮;DQ:发育商 EOEE:early onset epileptic encephalopathy;CNV-seq:copy number variations sequencing;ACMG:American College of Medical Genetics and Genomics;P:pathogenic;IS:infantile spasm;VPA:Valproate;TP:Topiramate;LEV:Levetiracetam;PB:Phenobarbital;CLP:Clonazepam;DQ:developmental quotient
基因与CNV是EOEE的重要潜在病因[4]。本组资料60例EOEE患儿基因诊断明确24例(40.0%),其中单基因变异20例(33.3%),CNV变异4例(6.7%),检测结果与既往报道基本符合[5];共检出13种单基因变异,其中15处变异位点和5种CNV变异既往国内外文献均未见报道。IS是婴幼儿期最常见的年龄特异性癫痫性脑病,本资料中10例IS患儿有3例存在TSC基因变异,例1同时存在TSC1、TSC2突变,胞姐是TSC1杂合变异,均遗传自父亲;其父仅面部有皮脂腺瘤,姐姐表型正常,同时存在TSC1、TSC2突变文献报道十分少见;3例患儿诊断结节性硬化症(TSC)临床标准均不足,均通过基因确诊,TSC是最常引起IS的神经皮肤疾病。
本研究中与IS相关的基因还有SCN8A、ZEB2、HNRNPU、DCX等,其中SCN8A基因以新发变异为主,极少数遗传,表型异质性强[6]。例9患儿9月龄时出现痉挛发作,起病前发育正常,起病后发育轻度落后,脑电图可见高峰失律,确诊IS,行全外显子联合CNV检测出系SCN8A基因杂合错义突变,遗传自有癫痫病史的父亲,其父幼年起病,语言表达稍差,智力大致正常,目前同时服用卡马西平和拉莫三嗪,偶有发作。
ZEB2基因突变可导致Mowat-Wilson综合征(MWS),表现为难治性癫痫、发育落后、特殊面容、多脏器畸形、先天性巨结肠症等[7];由于基因的大片段缺失、移码或无义变异导致单倍体剂量不足而致病,错义突变少见[8]。例15系ZEB2基因新生无义突变(c.2456C>G,p.Ser819X),表型为IS,ZEB2变异导致IS十分少见,本例是国内首次报道。
例20为HNRNPU移码突变(p.Arg224Glyfs*11),是23个碱基对缺失的结果,最终导致翻译提前终止。HNRNPU基因的大片段缺失与智力低下和癫痫发作相关,目前国内外报道大部分为染色体片段微缺失部分含有该基因缺失,资料中例23就检出2处CNV异常,即1q43-1q44缺失和Xp22.339重复,缺失区域包括EOEE的致病基因HNRNPU;患儿表现为精神发育迟滞、特殊面容、小角膜、胼胝体异常等,与文献报道[9,10]相符。HNRNPU基因点突变少见,几乎所有患儿均在出生1年内起病,多有热敏感性,表现为痉挛发作,大多早期有精神运动发育落后。
DCX为X连锁基因,此类基因变异对男女患者的影响不同。女性DCX基因变异表现为典型的双皮质综合征,男性则为无脑回畸形[11];女性患病与X染色体随机失活有关;男性表型更重,早期出现严重的认知、语言障碍和癫痫发作等症状[12]。例14系DCX基因错义突变女性患儿,表型为IS、皮质下层状灰质异位。资料中与X连锁相关的基因还有CDKL5、PCDH19,CDKL5基因变异女性患病率高,男女表型严重程度无明显差异[13]。例11女童系CDKL5基因新发移码突变(c.1993_1996del),出生1个月出现癫痫发作,发作频繁、多种发作形式,6个月时不会追光追物、不会抬头、仅发声。研究发现[13]CDKL5相关IS患儿接受促皮质素(ACTH)治疗后易出现频繁惊厥发作,此类患儿应慎用ACTH。
另外在诊断为非已知癫痫性脑病的患儿中发现存在PPP3CA基因突变,目前国内未见相关报道,该基因突变导致婴儿或儿童早期癫痫性脑病1型[14];例16系PPP3CA基因新发杂合移码缺失(c.1255_1256delAG),患儿2个月时出现癫痫发作,包括局灶性、阵挛性、全面性强直阵挛等多种发作形式,目前4岁,不会说,不会抬头,仅发声、没有语言,肌张力低,预后很差。例17系CACNA1E基因错义突变,表型为难治性癫痫、严重肌张力减退及发育落后。CACNA1E功能获得性变异是导致EOEE的主要原因[15],阻断R型钙电流是开发CACNA1E相关癫痫精准药物的一个有希望的靶点。
除外以上的单基因变异,还发现4例CNV变异患儿,主要表型包括精神运动发育迟滞、特殊的颅面特征、多脏器畸形、难治性癫痫等。例21系17p13.3缺失,内含LIS1等基因,LIS1缺失可导致Miller-Dieker综合征(MDS)[16],该患儿有发育落后、肌张力异常、无脑回畸形、IS等,符合MDS[16]。例22系11q23.3q25缺失,缺失区域中的FLI1基因参与血管和巨核细胞的分化,缺失可导致血小板减少,该患儿表现为发育落后、IS、血小板减少等;此综合征还可有癫痫发作,但表型为IS系首次报道[17];例23缺失区域包括EOEE的致病基因HNRNPU。例24系与1p36缺失综合征相关,此类缺失综合征多数有癫痫发作,以IS最常见[18]。该患儿表型为追光追物差、癫痫、手指畸形等。除病例23外,其余3例均缺乏典型的颅面部特征考虑与年龄小有关。
传统方法如临床症状、头颅MRI和遗传代谢检测等对癫痫性脑病的病因诊断率<13%,二代靶向测序可将诊断率提高至30%左右[19]。本研究联合全外显子和CNV对EOEE进行检测,基因变异阳性率33.3%,CNV阳性率6.7%,总阳性率达40.0%;联合CNV检测可提高EOEE病因的检测阳性率,与既往文献报道一致[5,19,20]。一项回顾性文献报道显示,癫痫性脑病患儿的起病年龄(27.23±44.05)个月(7 d~15岁),均通过基因检测确诊,确诊年龄(6.8±4.7)年(5个月~17岁)[19];本研究中患儿就诊年龄2 d~4岁(中位年龄10个月),就诊后立即进行基因检测,检测耗时20个工作日,患儿的确诊年龄可控制在就诊1个月以内,即1个月~4岁1个月(中位年龄11个月),确诊年龄明显小于上述报道年龄。另有研究显示,早期对EOEE患者进行基因测序,不仅可将诊断年龄提前,并有助于早期精准治疗、减少医疗支出,具有显著的临床效益和成本效益[21]。本研究连同上述文献报道为基因测序在EOEE早期诊断中的临床应用价值提供了有力的证据。
本研究对不明原因EOEE联合采用全外显子组和CNV-seq技术检测,丰富了EOEE的基因型,提高了EOEE的病因诊断水平;对这类患儿早期进行遗传学检测,可以早期确诊,避免不必要的后续检查并有助于癫痫精准治疗、判断预后及再生育指导。
所有作者均声明不存在利益冲突

























