
囊性纤维化(CF)是一种常染色体隐性单基因遗传病,由于囊性纤维化跨膜传导调节蛋白(CFTR)基因纯合和双重杂合突变所致。CFTR基因编码的是一种氯离子通道蛋白,分布于气道、胰腺导管上皮等部位,其功能异常将产生一系列临床表现。近10年来针对CFTR功能缺陷这一源头问题的特异性分子调节治疗取得了令人瞩目的成果。CF也因此成为一种模式疾病,为其他罕见的遗传疾病在遗传学、分子和细胞发病机制以及药物研发方面开创了新路线。现就近年来CF的发病机制以及分子调节治疗的最新研究进展进行综述,旨在提高广大医师对该病机制的认识,了解分子调节治疗。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
囊性纤维化(cystic fibrosis,CF)是高加索人群中最常见的致死性遗传性疾病,发病率为1/2 500~1/1 800,根据人口登记的估计,全世界目前有近9万人患有CF[1]。1938年由Dorothy Andersen首次报道。在20世纪四五十年代对CF患者的报道主要集中在由于胰腺损伤导致的严重吸收不良、消瘦和儿童期的高死亡率,同时也注意到了患者儿童期肺部感染的问题。在1948年纽约热浪期间,许多CF患儿出现了严重的低渗性脱水,由此发现了汗液失盐的情况,进而发展出汗液钠、氯离子检测的诊断方法。自从1989年囊性纤维化跨膜传导调节蛋白(CFTR)基因发现后,对该病发病机制的理解得到了更大提升,也逐渐发展了新的治疗方法。既往对CF的治疗重点在于控制临床症状,近年的治疗目标已经进展为针对病因进行治疗,也为其他遗传性疾病的治疗打开了新大门。以往认为CF在我国十分罕见,但近年来随着医疗水平的发展以及对该病认识的提高,我国CF诊断及报道的病例数明显升高,这提示了以往CF在中国的发病率可能被低估。现通过对CF的发病机制及分子调节治疗的进展进行文献综述,旨在提高广大临床医师对该病的认识,了解分子调节治疗。
CF是常染色体隐性遗传病,由于CFTR基因突变所致,其编码蛋白CFTR是一种环磷酸腺苷(cAMP)依赖性氯离子通道蛋白,由1 480个氨基酸构成,有2个亲水位点,各由6个镶嵌在细胞膜内的疏水环组成的跨膜区和2个重复的氨基酸结构,裂解ATP酶,为跨膜转运提供能量[2]。CFTR作为离子通道可以驱动上皮的氯离子和碳酸氢盐分泌,主要分布在汗腺、气道、胃肠道、胰腺和输精管。
在呼吸系统,CFTR除了介导气道上皮氯离子和碳酸氢盐分泌,还可以通过抑制上皮钠离子通道(ENaC)调节钠离子的吸收,另外对其他氯离子通道(ANO1、SLC26A9)和阴离子交换蛋白(SLC26A4)也有正向调节作用[3]。正常情况下气道表面上皮细胞可以通过表达ENaC和CFTR控制钠离子的吸收和氯离子的分泌,以调节气道表面液体的量和成分。而CF患者的气道表面液体明显减少,导致气道表面脱水,黏液脓性增加,严重影响黏液纤毛清除作用。同时碳酸氢盐分泌减少可降低气道表面液体pH值,影响乳铁蛋白及其他防御素的功能。由此造成气道内慢性感染及炎症形成恶性循环,进而破坏气道及肺结构,形成支气管扩张。
CFTR在胰腺导管细胞膜顶端高表达,是胰腺分泌氯离子、碳酸氢盐和液体必不可少的。碳酸氢盐可以中和胃酸,使消化液的pH值适于消化酶。CF患者由于CFTR功能异常,胰腺导管上皮受损及梗阻,血液中的胰腺蛋白(包括胰蛋白酶原)水平升高。免疫反应性胰蛋白酶原测定也是多数新生儿筛查系统的基础。持续的导管阻塞、炎症、纤维化和脂肪浸润最终导致胰腺的破坏。胰腺的破坏在胚胎期即发生,约85%的CF患者在出生时外分泌腺体已被完全破坏[3]。在胃肠道由于碳酸氢盐分泌不足造成肠黏液阻塞和胎粪性肠梗阻[4]。类似的情况也发生在肝脏,CF肝病的特征是胆汁分泌高黏,胆汁淤积,并最终导致肝硬化[5]。
汗腺分泌的汗液主要由水和盐组成,通常为低渗性,而CF患者由于CFTR功能丧失导致NaCl不能被正常回吸收,进而造成汗液中盐分过多丢失。由于CFTR在男性生殖系统(如附睾、精囊、输精管)和女性生殖系统(子宫和阴道)中均有表达。CF患者的生殖系统在胚胎发育过程中可能会出现畸形,男性可能会出现先天性双侧输精管缺失。另外碳酸氢盐与精子运动和卵子受精能力有关,因而CF患者的生育能力会受影响[3]。
临床表现与CFTR功能缺陷有关,不符合CF诊断标准的病例被定义为CFTR相关疾病。其临床表现局限于某一单独器官或系统,如单独出现的双侧输精管缺如、反复胰腺炎、弥漫性支气管扩张。这些疾病也与CFTR基因变异相关,对CFTR功能影响轻微。临床医师也应意识到CFTR基因变异可能会影响到更广泛的人群。
CFTR基因位于7号染色体长臂3区1带,全长约250 kb,共27个外显子。目前已发现约2 000种突变,包括错义突变(39%)、移码突变(16%)、剪切突变(11%)、无义突变(8%),大片段缺失或插入(2%)[6]。
尽管所有突变均会造成CFTR功能缺陷,但为了更好地进行分子结构-功能研究并为突变特异性矫正治疗提供科学依据,根据对CFTR功能影响的机制不同可将CFTR基因突变分为6类(表1[7,8]):第 Ⅰ 类突变:影响蛋白合成,包括大多数无义突变,导致mRNA降解;第 Ⅱ 类突变:影响蛋白运输,蛋白质因错误折叠在内质网中发生降解,而不能运输至细胞表面,从而严重影响CFTR功能;第 Ⅲ 类突变(门控突变):CFTR通道门控破坏,不能正常开启;第 Ⅳ 类突变:CFTR通道对氯离子和碳酸氢盐的传导性明显下降;第 Ⅴ 类突变:由于选择性剪接而同时产生正常和异常的mRNA,致使正常CFTR数量下降;第Ⅵ类突变:通过增加CFTR的内吞作用或减少其返回细胞表面,造成CFTR在细胞表面不稳定。第 Ⅰ~Ⅲ 类突变可导致CFTR功能丧失,当患者2个CFTR基因拷贝同时存在第 Ⅰ~Ⅲ 类突变时,表型严重,存在有胰腺功能不全。而第 Ⅳ~Ⅵ 类突变可有CFTR残余功能,通常表型轻。
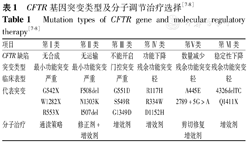
| 项目 | 第 Ⅰ 类 | 第 Ⅱ 类 | 第 Ⅲ 类 | 第 Ⅳ 类 | 第Ⅴ类 | 第 Ⅵ 类 |
|---|---|---|---|---|---|---|
| CFTR缺陷 | 无合成 | 无运输 | 不能开启 | 功能下降 | 数量减少 | 稳定性下降 |
| 突变类型 | 最小功能突变 | 最小功能突变 | 门控突变 | 残余功能突变 | 残余功能突变 | 残余功能突变 |
| 临床表型 | 严重 | 严重 | 严重 | 轻 | 轻 | 轻 |
| 代表突变 | G542X | F508del | G551D | R117H | A445E | 4326delTC |
| W1282X | N1303K | S549R | R334W | 2789+5G>A | Q1411X | |
| R553X | I507del | G1349D | D1152H | |||
| 分子治疗 | 通读策略 | 修正剂+增效剂 | 增效剂 | 增效剂 | 剪切修复增效剂 | 增效剂 |
随着可以改善CFTR功能的修正剂和增效剂的上市,为了更易于招募临床试验,近年来有学者提出将CFTR突变类型简单地分为两类,第一类是导致CFTR功能严重受损的突变,即最小功能突变,第二类是存在CFTR残余功能的突变[9]。
全球范围内,超过80%的CF患者存在至少1个等位基因携带F508del突变,其中约40%为纯合突变。在整个CF患者人群中,除F508del外只有5个突变(G542X、G551D、N1303K、R117H、W1282X)的患病率超过1%,约50个突变的患病率超过0.1%,而其他大多数突变是罕见的[7]。
近10年来针对CFTR功能缺陷这一源头问题的特异性分子调节治疗取得了令人瞩目的成果。CF也因此成为一种模式疾病,为其他罕见的遗传疾病在遗传学、分子和细胞发病机制以及药物研发方面开创了新路线。
CFTR增效剂依法卡托是第1种被成功研发的分子调节药物,可延长CFTR通道开放时间,加速细胞表面氯离子的转运,进而增强CFTR功能。最初的研究主要集中在G551D门控突变,这种突变的特点是细胞膜表面虽然有足够多的CFTR蛋白,但由于门控破坏,离子通道不能正常开启,进而几乎完全没有功能。首先体外试验证实依法卡托可使CFTR活性恢复约50%,并显著改善黏液纤毛清除率[10]。临床研究发现依法卡托可有效降低汗液氯离子浓度,改善肺功能,降低肺部病变急性加重次数,提高体重指数(BMI)及生活质量,其作用显著、迅速并且存在剂量依赖性和可逆性[11]。依法卡托于2012年获得美国食品药品监督管理局(FDA)批准上市,来自真实世界的注册研究同样显示依法卡托可有效降低CF患者肺部病变急性加重次数、改善肺功能、提高BMI、延缓CF相关糖尿病的发生[12]。尽管只有不到10%的CF患者可通过依法卡托治疗获益,但依法卡托的问世对于罕见病的精准治疗具有革命性的意义,也是后续发展的多重分子调节治疗的重要组成部分。
F508del是携带率最高的突变,属于第 Ⅱ 类突变,此类突变导致蛋白质错误折叠而被降解,不能被运输至细胞表面。CFTR修正剂通过不同的机制在内质网折叠过程中增强蛋白质构象的稳定性,从而起到挽救突变体的折叠、加工和运输[13]。第1代CFTR修正剂鲁玛卡托的上市让更多CF患者受益;3期临床试验显示鲁玛卡托与依法卡托联合使用可以改善F508del纯合突变患者肺功能水平[14]。由于鲁玛卡托的不良反应明显,耐受性更好的第2代CFTR修正剂替扎卡托被更多地用于临床实践[15]。但上述联合治疗对F508del纯合突变患者的作用不够明显。为了更好地改善CFTR在细胞内加工和转运缺陷,将CFTR功能提高到仅1个等位基因携带致病突变的水平以上,科学家研发了第3代CFTR修正剂,其可以通过结合到不同的CFTR区域为替扎卡托提供互补和附加的修正作用,即巴莫卡托和依来卡托。新一代CFTR修正剂与替扎卡托和依法卡托共同组成的三联疗法在体外试验和随机、双盲、安慰剂或活性药物对照的2期临床试验均证实对基因型为F508del/最小功能突变及F508del纯合突变患者有效[16,17]。2019年发表的一项3期临床试验结果显示三联疗法可以明显改善F508del纯合突变患者的肺功能[18]。2021年发表的1篇3期临床试验进一步证实了三联疗法对于携带F508del突变患者的有效性[19]。这项研究在纳入患者的基因型上选择了F508del/门控突变或F508del/残余功能突变,导入期及对照组采用活性药物,即依法卡托或替扎卡托-依法卡托治疗(均已证实对门控或残余功能突变有效),这样的试验设计进一步证实了三联疗法主要增强F508del等位基因的活性,可以为具有单个F508del突变加上一个对原有CFTR调节剂有响应的门控或残余功能突变的患者提供额外的好处[19]。半数患者在经过8周三联治疗后汗液氯离子浓度降至30 mmol/L以下,即达到了健康人汗液氯离子浓度[18,19]。此外,上述研究均显示三联疗法的安全性及耐受性良好[16,17,18,19]。三联疗法扩大了CFTR调节治疗的有效范围,有望帮助70%~90%的CF患者。
通读策略可通过解除反义过程中因终止密码子(PTC)导致的"解码错误"而恢复蛋白质的正常合成,理论上适用于所有无义突变所致疾病。第 Ⅰ 类突变由于PTC的插入导致mRNA降解,进而影响CFTR合成。体外试验发现ELX-02(NB124)可恢复G542X、R553X、R1162X、W1282X 4种最常见的PTC突变体的部分CFTR功能,如与CFTR增效剂联合使用效果将更为显著[20]。在健康志愿者参与的 Ⅰ 期临床试验中,尽管有轻微的不良反应发生,但是EXL-02具有良好的耐受性和安全性[21]。目前正在进行的多项临床试验评估ELX-02剂量递增对至少1个等位基因携带G542X突变的CF患者的影响(NCT04126473,NCT04135495)[22]。
除了前述针对CFTR本身的分子调节治疗外,还有研究作用于其他离子通道,以从旁路纠正CFTR功能缺失所导致的渗透压和酸碱失衡,即旁路治疗。通过激活其他氯离子通道,如ANO1和ANO6,可以替代CFTR下降的功能。ENaC抑制剂(苯扎米尔和阿米洛利)可通过抑制钠离子内流改善CFTR功能缺失导致的钠离子高吸收情况。理论上旁路治疗可以适用于各种类型突变,但目前尚不能证实其在临床中的安全性和有效性[23]。
以往认为CF在我国十分罕见。近年来随着对CF认识水平的提高和分子诊断技术的进步,国内诊断CF病例逐渐增多。我国CF患者常见的突变类型包括G970D、c.1766+5G>T、I1023R,大多数突变类型在白种人中罕见或未见,而F508del突变报道很少[24]。虽然CFTR分子治疗飞速发展,但是目前尚无证据表明上述分子调节治疗可用于我国患者,这需要对国人CFTR突变基因更全面的了解,以及进一步的基础与临床研究。随着国内诊断患者增多和多中心注册研究的开展,相信在不久的将来,分子调节治疗也将给中国CF患者带来福音。
所有作者均声明不存在利益冲突

























