
分析常染色体显性遗传Alport综合征(ADAS)患儿的临床病理和基因突变特征,以提高对其的认识。
纳入2016年9月至2020年2月在华中科技大学同济医学院附属同济医院儿科诊断为ADAS的10例患儿,回顾性分析其临床病理特点及基因突变特征,并随访。
1.ADAS患儿中位诊断年龄5.7(2.4,9.8)岁,6例(60.0%)患儿有肾衰竭家族史,4例(40.0%)患儿确诊时表现为血尿合并蛋白尿,2例(20.0%)患儿出现高频听力受损。肾脏活检显示4例(40.0%)患儿肾小球基底膜(GBM)致密层为广泛撕裂分层样改变,6例(60.0%)患儿为局限性撕裂病变。2.10例患儿中4例(40.0%)为COL4A3基因杂合突变,2例为甘氨酸点突变,2例为剪接突变;6例(60.0%)患儿为COL4A4基因杂合突变,其中4例为胶原区甘氨酸点突变,1例为无义突变,1例为大片段缺失。上述突变中6个为未见报道的新突变。
ADAS患儿早期临床表现多不典型,肾外表现较少见,GBM致密层以局限性撕裂分层为主,基因突变以甘氨酸点突变为主。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Alport综合征(AS)是罕见的遗传性进行性肾小球肾炎,以进行性肾功能减低、锥形晶状体和感音神经性耳聋为主要临床表现[1]。基底膜 Ⅳ 型胶原基因突变是导致AS的原因,包括COL4A5、COL4A4和COL4A3[2]。AS存在3种遗传方式,80%为COL4A5基因突变导致的X连锁显性遗传,15%为COL4A4、COL4A3基因突变导致的常染色体隐性遗传[3],常染色体显性遗传Alport综合征(ADAS)最罕见,表现不典型,不易识别。国际上报道较少,国内仅见个例报道[4,5,6,7]。本研究报道了来自9个家系的10例ADAS患儿的临床表现及病理特点,以提高对ADAS的认识。
回顾性研究。病例来源为2016年9月至2020年2月在华中科技大学同济医学院附属同济医院儿科住院诊断为ADAS的患儿。纳入标准[8]:(1) 符合AS诊断标准:患儿存在血尿或血尿合并蛋白尿等肾小球肾炎表现,有或无肾衰竭家族史,肾脏病理表现为电镜下肾小球基底膜(GBM)致密层撕裂分层改变;(2) 家系符合常染色体显性遗传特征或经基因检测结果判定为常染色体显性遗传;(3)诊断时年龄<18岁。共纳入10例患儿。排除标准:不符合AS诊断,遗传方式为X连锁显性遗传、常染色体隐性遗传或双基因遗传。本研究已通过医院医学伦理委员会批准(批准文号:TJ-IRB20211103),已豁免监护人知情同意。
患儿均进行完整临床检查和评估,包括一般资料、临床表现、家族史,尿常规、尿白蛋白/肌酐比值、24 h尿蛋白定量、血生化和血清学检查。病理资料包括肾脏组织病理光镜、电镜、免疫荧光[免疫球蛋白(Ig)A、IgG、IgM、补体C3、补体C1q]、Ⅳ型胶原α3/α5链免疫荧光染色结果,抗Ⅳ型胶原α3/α5链单克隆抗体(瑞典WiesLab)。
采集先证者及其家系成员外周血2~3 mL,全外显子组测序服务由深圳华大基因科技有限公司和上海明码生物科技有限公司提供。取300 ng基因组DNA利用片段化酶打断,磁珠筛选后获得平均大小为200~300 bp的DNA片段,经末端修复、接头连接和聚合酶链式反应进行文库构建,文库与定制的探针进行杂交,杂交结束后进行探针的洗脱与洗涤,洗脱后杂交文库用MGISEQ-2000测序平台进行高通量测序,测序原始数据使用BWA软件与hg19人类参考基因组进行序列比对,采用GATK软件进行插入缺失、单核苷酸多态性位点等分析,在HapMap数据库、千人基因组数据库、dbSNP等数据库对原始数据进行注释,结合患者临床资料和生物信息学软件预测结果筛选突变基因,并进行Sanger测序验证。根据美国医学遗传学与基因组学学会(ACMG)遗传变异分类标准与指南[9]对变异的致病性进行判读。
应用SPSS 26.0软件进行统计学分析,计数资料用百分比表示,计量资料用M(P25,P75)表示。
来自9个家系的10例ADAS患儿的临床特征见表1。其中男4例(40.0%),女6例(60.0%),中位诊断年龄5.7(2.4,9.8)岁,<10岁诊断为AS的有8例(80.0%)。6例(60.0%)患儿有肾衰竭家族史,家系成员中肾衰竭时中位年龄为40.5岁。4例(40.0%)确诊时肾脏表现为血尿合并蛋白尿,病例7在9岁首次就诊时出现大量蛋白尿。
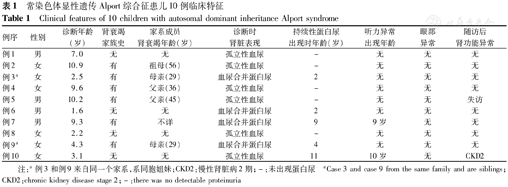
常染色体显性遗传Alport综合征患儿10例临床特征
Clinical features of 10 children with autosomal dominant inheritance Alport syndrome
常染色体显性遗传Alport综合征患儿10例临床特征
Clinical features of 10 children with autosomal dominant inheritance Alport syndrome
| 例序 | 性别 | 诊断年龄(岁) | 肾衰竭家族史 | 家系成员肾衰竭年龄(岁) | 诊断时肾脏表现 | 持续性蛋白尿出现时年龄(岁) | 听力异常出现年龄 | 眼部异常 | 随访后肾功能异常 |
|---|---|---|---|---|---|---|---|---|---|
| 例1 | 男 | 7.0 | 无 | 无 | 孤立性血尿 | - | 无 | 无 | 无 |
| 例2 | 女 | 10.9 | 有 | 祖母(56) | 孤立性血尿 | - | 无 | 无 | 无 |
| 例3a | 女 | 2.5 | 有 | 母亲(29) | 血尿合并蛋白尿 | 2 | 无 | 无 | 无 |
| 例4 | 女 | 9.6 | 有 | 父亲(36) | 孤立性血尿 | - | 无 | 无 | 无 |
| 例5 | 男 | 10.2 | 有 | 父亲(45) | 孤立性血尿 | - | 无 | 无 | 失访 |
| 例6 | 男 | 1.6 | 无 | 无 | 血尿合并蛋白尿 | 2 | 无 | 无 | 无 |
| 例7 | 男 | 9.3 | 有 | 不详 | 血尿合并蛋白尿 | 9 | 9岁 | 无 | 无 |
| 例8 | 女 | 2.2 | 无 | 无 | 孤立性血尿 | - | 无 | 无 | 无 |
| 例9a | 女 | 4.3 | 有 | 母亲(29) | 血尿合并蛋白尿 | 4 | 无 | 无 | 无 |
| 例10 | 女 | 3.1 | 无 | 无 | 孤立性血尿 | 11 | 10岁 | 无 | CKD2 |
注:a例3和例9来自同一个家系,系同胞姐妹;CKD2:慢性肾脏病2期;-:未出现蛋白尿 aCase 3 and case 9 from the same family and are siblings;CKD2:chronic kidney disease stage 2;-:there was no detectable proteinuria
随访9例,失访1例,随访时间1.7(0.9,2.8)年,随访过程中8例患儿未出现肾功能异常,病例8由孤立性血尿发展为血尿合并蛋白尿,病例10在12岁时进展至慢性肾脏病2期(CKD2),并出现大量蛋白尿,家系图谱见图1。
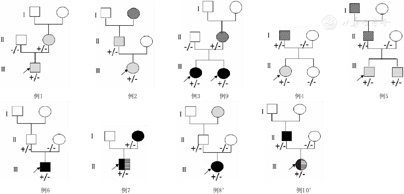
注:□:男性;○:女性;白色:无临床表现;灰色:孤立性血尿;黑色:血尿合并蛋白尿;横线:肾外表现;斜线:肾衰竭; :先证者;-:野生型等位基因;+:突变体等位基因;a例8、例10诊断时肾脏表现为孤立性血尿,随访后出现蛋白尿 □:male;○:female;white symbols:individuals without clinical manifestations;filled grey symbols:individuals with isolated hematuria;filled black symbols:individuals with hematuria and proteinuria;horizontal lines:individuals with extrarenal manifestations;oblique line:individuals with renal failure;
:先证者;-:野生型等位基因;+:突变体等位基因;a例8、例10诊断时肾脏表现为孤立性血尿,随访后出现蛋白尿 □:male;○:female;white symbols:individuals without clinical manifestations;filled grey symbols:individuals with isolated hematuria;filled black symbols:individuals with hematuria and proteinuria;horizontal lines:individuals with extrarenal manifestations;oblique line:individuals with renal failure; :the proband;-:wild type allele;+:mutated allele;acases 8 and case 10 presented isolated hematuria at diagnosis and developed proteinuria during the follow-up
:the proband;-:wild type allele;+:mutated allele;acases 8 and case 10 presented isolated hematuria at diagnosis and developed proteinuria during the follow-up
10例患儿肾脏活检结果见表2。光镜以轻微病变为主,免疫荧光大多未见免疫复合物和补体沉积。4例(40.0%)患儿GBM致密层为典型撕裂分层样改变,6例(60.0%)患儿为局限性撕裂病变。
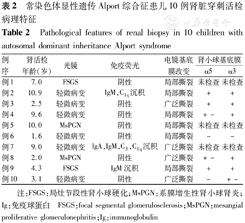
常染色体显性遗传Alport综合征患儿10例肾脏穿刺活检病理特征
Pathological features of renal biopsy in 10 children with autosomal dominant inheritance Alport syndrome
常染色体显性遗传Alport综合征患儿10例肾脏穿刺活检病理特征
Pathological features of renal biopsy in 10 children with autosomal dominant inheritance Alport syndrome
| 例序 | 肾活检年龄(岁) | 光镜 | 免疫荧光 | 电镜基底膜改变 | 肾小球基底膜 | |
|---|---|---|---|---|---|---|
| α5 | α3 | |||||
| 例1 | 7.0 | FSGS | 阴性 | 局部撕裂 | 未检查 | 未检查 |
| 例2 | 10.9 | 轻微病变 | IgM,C1q沉积 | 局部撕裂 | + | + |
| 例3 | 2.5 | 轻微病变 | 阴性 | 广泛撕裂 | + | + |
| 例4 | 9.6 | 轻微病变 | 阴性 | 局部撕裂 | +- | + |
| 例5 | 10.0 | MsPGN | 阴性 | 局部撕裂 | 未检查 | 未检查 |
| 例6 | 1.6 | 轻微病变 | 阴性 | 局部撕裂 | - | - |
| 例7 | 9.0 | 轻微病变 | IgA,IgM,C3,C1q沉积 | 广泛撕裂 | 未检查 | 未检查 |
| 例8 | 2.0 | MsPGN | 阴性 | 广泛撕裂 | +- | + |
| 例9 | 4.3 | FSGS | IgM沉积 | 局部撕裂 | + | + |
| 例10 | 3.1 | 轻微病变 | 阴性 | 广泛撕裂 | - | +- |
注:FSGS:局灶节段性肾小球硬化;MsPGN:系膜增生性肾小球肾炎;Ig:免疫球蛋白 FSGS:focal segmental glomerulosclerosis;MsPGN:mesangial proliferative glomerulonephritis;Ig:immunoglobulin
7例患儿进行了Ⅳ型胶原纤维α3、α5免疫组织化学检测,其中3例GBM α3和α5表达正常,1例α3表达减弱且α5未见表达,2例α3表达正常α5表达减弱,1例α3和α5均无表达。
10例患儿检测到的突变结果见表3[10,11,12,13],其中4例(40.0%)为COL4A3基因杂合突变,6例(60.0%)为COL4A4基因杂合突变。主要突变类型为胶原区甘氨酸取代(6/10例),其次为剪接突变(2/10例)。剪接突变均出现在COL4A3基因,且临床表现较重,1例出现肾功能减低(CKD2期),1例表现为血尿和大量蛋白尿。检索ClinVar数据库和The Human Gene Mutation Database,发现6种突变为未见报道的新突变,根据ACMG遗传变异分类标准指南,评价6种突变为可能致病性。
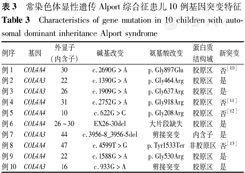
常染色体显性遗传Alport综合征患儿10例基因突变特征
Characteristics of gene mutation in 10 children with autosomal dominant inheritance Alport syndrome
常染色体显性遗传Alport综合征患儿10例基因突变特征
Characteristics of gene mutation in 10 children with autosomal dominant inheritance Alport syndrome
| 例序 | 基因 | 外显子(内含子) | 碱基改变 | 氨基酸改变 | 蛋白质结构域 | 新突变 |
|---|---|---|---|---|---|---|
| 例1 | COL4A4 | 30 | c.2690G>A | p.Gly897Glu | 胶原区 | 否[10] |
| 例2 | COL4A3 | 22 | c.1390G>A | p.Gly464Arg | 胶原区 | 是 |
| 例3 | COL4A3 | 26 | c.1909G>A | p.Gly637Arg | 胶原区 | 是 |
| 例4 | COL4A4 | 31 | c.2752G>A | p.Gly918Arg | 胶原区 | 否[11] |
| 例5 | COL4A4 | 10 | c.622G>C | p.Gly208Arg | 胶原区 | 否[12] |
| 例6 | COL4A4 | 26~30 | EX26-30del | 大片段缺失 | 胶原区 | 是 |
| 例7 | COL4A3 | 44 | c.3956-8_3956-5del | 剪接突变 | 内含子 | 是 |
| 例8 | COL4A4 | 47 | c.4599T>G | p.Tyr1533Ter | 非胶原区 | 否[13] |
| 例9 | COL4A4 | 22 | c.1588G>A | p.Gly530Arg | 胶原区 | 是 |
| 例10 | COL4A3 | 16 | c.933G>A | 剪接突变 | 胶原区 | 是 |
AS的发病率为1/10 000~1/5 000[14],以X连锁显性遗传为主。临床表现中男女区别较大,X连锁遗传男性较为严重,70%的患者在30岁时发生肾衰竭[15]。常染色体隐性遗传AS患者在21岁左右时发生肾衰竭[16]。ADAS在AS中非常罕见,近几年的报道正在逐渐增多[8,17,18],但ADAS在儿童中报道较为少见。本文对来自9个家系的10例ADAS患儿的临床病理表现及基因检测结果进行总结分析,其中位诊断年龄为5.7岁。有报道ADAS发病年龄较晚,在29岁左右[17]。Marcocci等[19]报道的38例ADAS患者中50%存在蛋白尿,本研究中40%的患者首次就诊时即存在持续性蛋白尿。6个家系成员存在肾衰竭史,肾衰竭时中位年龄为40.5岁,较文献报道的ADAS肾衰竭时的中位年龄小。Kamiyoshi等[8]报道的72例ADAS患者中位肾衰竭年龄为70.0岁,本研究与文献报道存在一定差异,可能与本研究中肾衰竭例数较少有关。
ADAS肾外表现发生率报道不一。与成人相比,儿童病例肾外表现较少见。在本研究中未发现有眼部病变的患儿,仅有2例患儿在9岁和10岁时出现听力受损。Rosado等[17]报道的听力损伤及眼部病变分别为68.4%、15.8%,Furlano等[18]报道的听力及眼部病变发生率分别为9.0%及0.8%,不同文献报道数据相差较大。除此MYH9基因变异也可引起肾脏受损、耳聋等临床表现,应注意与AS相鉴别[20]。
肾活检电镜检查对区分AS和薄基膜肾病(TBMN)有重要价值。在TBMN中,GBM弥漫变薄。AS的GBM则表现为广泛的撕裂分层及网状改变。ADAS与TBMN在临床表现上也存在差异,AS患儿往往存在肾衰竭家族史,临床表现有显著蛋白尿和肾功能进行性减低,肾外表现较常见,这些特点在TBMN患者中很少见[21]。本研究4例(40.0%)患儿的电镜下GBM表现为典型撕裂分层改变,6例(60.0%)表现为GBM的变薄及节段性撕裂分层。Kamiyoshi等[8]报道的16例ADAS患者中9例(56.3%)GBM有网状改变,Mastrangelo等[22]报道17例ADAS患者中71%的患者GBM有典型改变,本研究中出现GBM典型改变的例数较少,可能与其肾脏穿刺时年龄较小有关。ADAS患者中出现典型GBM改变时的年龄较晚,因此,年幼儿童肾活检GBM改变不典型,价值有限,难以作为诊断依据[23]。对于早期电镜下基底膜广泛变薄,有不典型撕裂分层并发现存在基因COL4A4或COL4A3杂合突变的儿童,规律定期随访很重要。
基因检测是诊断AS的重要手段[24]。本研究中4例存在COL4A3基因杂合突变,6例为COL4A4基因杂合突变,以COL4A4和COL4A3基因甘氨酸点突变为主,与既往报道一致[18]。6种突变为未见报道的新突变,其中3种突变位于COL4A3、COL4A4基因编码的三螺旋Gly-X-Y(X,Y为其他氨基酸)三肽重复结构域上,以及1种大片段缺失,软件进行蛋白功能预测,结果均为有害。2种剪接突变软件预测其可能影响蛋白的正常剪接,该突变的致病性有待进一步的功能验证。
本研究2例患儿(例6、8)表现为血尿合并蛋白尿,肾活检有明显AS改变,其突变基因均来源于父亲,但2例患儿的父亲未出现任何临床表现,可能原因:(1)部分ADAS家系显性不全;(2) 表观遗传修饰影响。也有类似报道,相同的致病变异导致不同的表型[8,25]。
临床上AS无特殊治疗药物,常用血管紧张素转化酶抑制剂及血管紧张素受体拮抗剂类药物控制蛋白尿,有研究显示早期应用这两类药物可有效延缓肾功能的恶化[26]。本研究中患儿出现蛋白尿例数较少,且随访时间相对较短,因此未进行药物相关分析。本研究为回顾性研究,具有一定的局限性,因研究对象样本数量较少且随访时间不够长,并未发现基因突变与临床表型之间有明显的相关性。
总之,ADAS患儿早期临床表现多不典型,肾外表现较少见,GBM致密层以局限性撕裂分层为主,基因突变以COL4A4或COL4A3胶原区甘氨酸点突变为主。肾衰家族史、GBM致密层撕裂分层以及基因突变分析是诊断的主要依据。
所有作者均声明不存在利益冲突

























