
对比分析济南地区甲基丙二酸血症(MMA)生化筛查与基因筛查的发病率及基因变异情况,调查济南地区人群中MMA相关致病基因的携带情况。
回顾性研究。统计2011年5月至2022年5月济南市新生儿疾病筛查中心通过串联质谱筛查确诊MMA的患儿,并对基因检测结果进行分析总结。收集6 800例新生儿的足跟血滤纸干血斑完成新生儿基因筛查,其中4 800例用高通量测序+目标区域捕获技术检测MMAA、MMAB、MMACHC及MMUT基因,2 000例采用超多重聚合酶链反应+目标基因位点捕获技术检测MMA相关8个基因174个目标基因位点。分析MMA的热点突变及相关基因携带率。
共367 452名新生儿完成串联质谱筛查,筛查确诊MMA患儿103例(男56例,女47例),MMA生物化学筛查的发病率为1∶3 567。其中76例获得基因确诊,检出4种MMA相关基因(MMACHC、MMUT、MMAA、MMADHC)变异。6 800例新生儿完成了新生儿基因筛查,共3例患儿确诊MMA,318例患儿携带甲基丙二酸的致病变异,总携带率为4.68%,其中MMACHC基因变异的携带率为3.09%(210/6 800),MMUT基因变异的携带率为1.43%(97/6 800)。
MMA为我国最高发的有机酸代谢障碍,济南地区该病发病率及携带率较高,新生儿基因筛查可作为新生儿生化筛查的重要补充,建议本地域育龄夫妇进行MMA相关致病基因的携带者筛查。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
甲基丙二酸血症(MMA)是我国最常见的有机酸代谢障碍性疾病,属常染色体隐性遗传病。主要由甲基丙二酰辅酶A变位酶自身缺陷(mut型)或其辅酶钴胺素代谢缺陷(cbl型)导致甲基丙二酸、甲基枸橼酸等代谢物异常蓄积引起,可导致多脏器及系统的功能损害[1,2,3,4,5]。根据是否合并同型半胱氨酸(Hcy)的增高可分为单纯型MMA和合并型MMA。在我国,由于串联质谱筛查技术的逐渐普及,近年来筛查确诊的MMA患儿逐渐增多,发现MMA的发病率总体较高且有明显的地域差异,目前发病率最高的报道是根据2011年至2014年期间济南市妇幼保健院筛查的35 291例新生儿得出的1∶3 920[6]。由于该病有晚发型患者,故生化筛查法时有漏筛漏诊的情况发生。目前对于已确诊MMA患儿基因变异的研究较多,但对人群中MMA基因变异的携带率尚不清楚。因此,本研究通过大样本量的筛查明确济南地区MMA的发病率,并进一步分析MMA患儿的基因型,对比分析MMA新生儿生化筛查与基因筛查的结果,了解人群中MMA相关基因的热点致病变异情况。
回顾性研究。选取2011年5月至2022年5月济南市新生儿疾病筛查中心通过串联质谱检测进行筛查的367 452例新生儿及2020年8月至2022年1月完成新生儿基因筛查的6 800例新生儿作为研究对象,其中6 800例接受基因筛查的新生儿均同步完成上述串联质谱筛查,血片采集与新生儿疾病筛查同步进行。本研究通过济南市妇幼保健院医院医学伦理委员会批准(批准文号:2020-1-028、2021-1-054),所有样本及医疗数据均已获得新生儿监护人签署的知情同意书。
新生儿出生72 h后并充分哺乳6次以上,用750 mL/L乙醇在新生儿足跟内侧或外侧消毒并挥发后用一次性采血针刺破皮肤,深度<3 mm,用干棉棒拭去第1滴血,从第2滴开始取样。将滤纸片接触血滴,使血自然渗透至滤纸背面,且每个血斑直径>8 mm,取至少3个血斑,血片自然晾干放到密封袋置于2~8 ℃保存。
采用美国Waters TQD串联质谱仪和NeoBaseT MSMS Kit(PerkinElmer公司)检测干血滤纸片中的丙酰基肉碱(C3)、乙酰基肉碱(C2)、蛋氨酸(Met)等指标,并计算C3与C2的比值。若C3、C3/C2升高,需召回复查,并留取患儿尿液10 mL,采用气相色谱法-质谱法尿气相质谱仪检测尿液中甲基丙二酸、甲基枸橼酸、3-羟基丙酸等有机酸的浓度;采用循环酶法检测血清中Hcy的浓度;结合血常规、血糖、肝肾功、心肌酶、血氨、头颅磁共振成像(MRI)等评估脏器功能。检测新生儿母亲血常规及叶酸、维生素B12水平,排除母源性MMA。将外周血或干血片寄送至北京迈基诺或杭州博圣公司进行基因检测。根据患儿临床表现(部分患儿可无症状),C3/C2、C3增高,尿甲基丙二酸、甲基枸橼酸增高,结合MMA相关基因分析可明确诊断。合并Hcy升高的诊断为合并型MMA,无Hcy升高的为单纯型MMA。对筛查确诊的MMA患儿进行治疗并定期随访。
(1)高通量测序+目标捕获技术:4 800例新生儿足底血干血滤纸片行新生儿基因筛查,需要血量为6个3.2 mm血斑,通过标准流程提取基因组DNA,再利用二代测序技术对致病基因的所有编码区进行捕获测序,核基因组有效测序深度≥100 X,线粒体基因有效测序深度≥300 X,目标区域的20 X覆盖度≥95%。对获得的原始测序数据进行拆分、比对和质控后,使用GATK软件进行单核苷酸变异/插入和缺失的检测。检测的MMA相关基因包括MMAA、MMAB、MMACHC及MMUT。检测结果只报道已知明确致病及疑似致病位点。(2)超多重聚合酶链反应(PCR)+目标基因位点捕获技术:2 000例新生儿足底血干血滤纸片行新生儿基因筛查,需要血量为4个3 mm血斑,使用超多重PCR技术对MMA的8个基因174个目标基因位点进行捕获和富集,选择的目标基因位点均为致病或疑似致病位点,分别为MMAA(25个基因位点)、MMAB(15个位点)、MMUT(90个位点)、MMADHC(13个位点)、MMACHC(16个位点)、LMBRD1(4个位点)、ABCD4(5个位点)、HCFC1(6个位点),富集的目的片段使用高通量测序平台进行测序,测序获得的DNA序列与人类基因组hg19参考序列进行比对并对目标区域的覆盖度和测序质量进行评估,对变异进行生物信息学分析和致病性分析。基因筛查有2个可疑致病或致病变异判读为筛查阳性,需召回进行父母的基因验证并结合生化筛查结果进入诊断程序,诊断程序同上。
进行疾病筛查的新生儿共367 452例。初筛阳性患儿9 494例,确诊MMA患儿103例(男56例,女47例),其中合并型MMA患儿88例(85.44%)、单纯型MMA患儿15例(14.56%)。济南地区MMA总发病率为1∶3 567,其中合并型MMA发病率为1∶4 176,单纯型MMA发病率为1∶244 967。串联筛查阳性的可疑患儿共82例完成了基因检测,其中76例检测到来自父母的2个致病变异,基因诊断明确,2例仅查到1个基因突变,4例未检测到MMA基因变异。未获得基因确诊的6例早期筛查指标C3/C2、C3及尿甲基丙二酸均轻度升高,予维生素B12治疗后代谢指标恢复正常,予停药随访,代谢指标维持正常,排除诊断。共检出4种基因(MMACHC、MUT、MMAA、MMADHC)的43种致病变异,其中MMACHC 23种、MMUT 17种、MMAA 2种、MMADHC 1种。常见的基因变异为MMACHC基因(82.58%)和MMUT基因(15.48%)。MMACHC基因常见的热点突变为c.609G>A(33.59%)、c.482G>A(14.84%)、c.80A>G(11.72%)、c.658_660delAAG(10.16%)、c.567dupT(4.69%)等;MMUT基因常见热点突变为c.1106G>A(12.50%)、c.1663G>A(12.50%)、c.914T>C(8.33%)、c.1963C>T(8.33%)、c.729_730insTT(8.33%)等,见表1。未做基因检测的患儿生化指标显著升高,临床诊断明确,并长期治疗随访。

新生儿4种甲基丙二酸血症相关基因变异类型分析
Analysis of the variation types of four methylmalonic acidemia related genes in neonates
新生儿4种甲基丙二酸血症相关基因变异类型分析
Analysis of the variation types of four methylmalonic acidemia related genes in neonates
| 基因 | 碱基改变 | 氨基酸改变 | 致病等级 | 频数(%) |
|---|---|---|---|---|
| MMACHC | c.609G>A | p.W203* | 致病变异 | 43(33.59) |
| c.482G>A | p.R161Q | 致病变异 | 19(14.84) | |
| c.80A>G | p.Q27R | 致病变异 | 15(11.72) | |
| c.658_660delAAG | p.K220del | 致病变异 | 13(10.16) | |
| c.567dupT | I190YfsX13 | 致病变异 | 6(4.69) | |
| c.656_658delAGA | p.219-220delQKinsQ | 致病变异 | 5(3.91) | |
| c.481C>T | p.R161* | 致病变异 | 4(3.13) | |
| c.394C>T | p.R132* | 致病变异 | 3(2.34) | |
| c.1A>G | p.M1? | 致病变异 | 3(2.34) | |
| c.445_446insA | p.C149X | 致病变异 | 2(1.56) | |
| c.445_446delTG | p.Cys149Hisfs*32 | 疑似致病 | 2(1.56) | |
| c.321G>A | p.V107V | 意义未明 | 1(0.78) | |
| c.626dupT | p.T210Dfs*35 | 致病变异 | 2(1.56) | |
| c.361G>T | p.A121S | 意义未明 | 1(0.78) | |
| c.275_276delAG | p.S93Pfs*11 | 疑似致病变异 | 1(0.78) | |
| c.217C>T | p.R73* | 致病变异 | 1(0.78) | |
| c.624_625del | p.V209Dfs*34 | 致病变异 | 1(0.78) | |
| c.228_231delTGAC | p.D77Qfs*22 | 疑似致病变异 | 1(0.78) | |
| c.452A>G | p.H151R | 意义未明 | 1(0.78) | |
| c.277-2_303del | / | 疑似致病变异 | 1(0.78) | |
| c.365A>T | p.H122L | 疑似致病变异 | 1(0.78) | |
| c.449T>G | p.I150R | 疑似致病变异 | 1(0.78) | |
| / | / | 致病变异 | 1(0.78) | |
| MMUT | c.1106G>A | p.R369H | 致病变异 | 3(12.50) |
| c.1663G>A | p.Ala555Thr | 致病变异 | 3(12.50) | |
| c.914T>C | p.Leu305Ser | 致病变异 | 2(8.33) | |
| c.1963C>T | p.R655C | 意义未明 | 2(8.33) | |
| c.729_730insTT | p.Asp244Leufs*39 | 致病变异 | 2(8.33) | |
| c.494A>G | p.D165G | 疑似致病变异 | 1(4.17) | |
| c.1630_1631delGGinsTA | p.G544X | 致病变异 | 1(4.17) | |
| c.613G>A | p.E205K | 疑似致病变异 | 1(4.17) | |
| c.755dupA | p.His252Glnfs*6 | 致病变异 | 1(4.17) | |
| c.599T>C | p.I200T | 意义未明 | 1(4.17) | |
| c.1286A>G | p.Y429C | 意义未明 | 1(4.17) | |
| c.1208G>A | p.Arg403Gln | 意义未明 | 1(4.17) | |
| c.278G>A | R39H | 致病变异 | 1(4.17) | |
| c.1084-10A>G | / | 疑似致病变异 | 1(4.17) | |
| c.1677-1G>A | / | 致病变异 | 1(4.17) | |
| c.2131G>A | p.E711K | 意义未明 | 1(4.17) | |
| c.460C>T | p.R154C | 意义未明 | 1(4.17) | |
| MMAA | c.650T>A | p.L217* | 致病变异 | 1(50.00) |
| c.563-524_*1627del | / | 致病变异 | 1(50.00) | |
| MMADHC | c.478+12T>C | / | 意义未明 | 1(100.00) |
注:/:内含子变异,该变异不影响氨基酸改变,仅影响剪切 /:intron variation,which does not affect amino acid changes,but only affects splicing
6 800例新生儿基因筛查结果显示,共3例确诊MMA,基因筛查发病率为1∶2 267,高于生化筛查的发病率(1∶3 567)。共318例患儿携带甲基丙二酸的致病变异,总携带率为4.68%,其中MMACHC基因变异的携带率为3.09%(210/6 800),MMUT基因变异的携带率为1.43%(97/6 800)。
4 800例新生儿通过该技术对MMA相关的4个基因(MMACHC、MMUT、MMAA、MMAB)进行筛查,其中MMACHC基因检测到23种致病突变,MMUT基因检测到29种致病突变,MMAA基因检测到3种致病突变,MMAB基因检测到5种致病突变。基因筛查确诊MMA患儿2例,分别为c.80A>G/c.609G>A及c.80A>G/c.482G>A的复合杂合突变。该2例患儿串联筛查结果均提示C3、C3/C2升高,召回复查生化结果符合合并型MMA诊断。未确诊的新生儿共251例携带MMA的致病基因,携带率为5.23%,其中163例携带MMACHC基因,携带率为3.40%,78例携带MMUT基因,携带率为1.63%,5例携带MMAA基因,携带率为0.10%,5例携带MMAB基因,携带率为0.10%。MMACHC基因的热点突变为c.609G>A(25.77%)、c.658_660delAAG(20.86%)、c.482G>A(13.50%)、c.80A>G(8.59%)、c.565C>T(6.13%)等;MMUT基因的热点突变为c.1663G>A(15.38%)、c.1208 G>A (15.38%)、c.729_730insTT(11.54%)、c.1106G>A(10.26%)、c.914T>C(7.69%)等。各筛查位点的携带情况见表2。

新生儿甲基丙二酸血症4 798例相关基因筛查携带情况
Carrying status of methylmalonic acidemia related gene screening in 4 798 neonates
新生儿甲基丙二酸血症4 798例相关基因筛查携带情况
Carrying status of methylmalonic acidemia related gene screening in 4 798 neonates
| 基因 | 碱基改变 | 氨基酸改变 | 频数(%) |
|---|---|---|---|
| MMACHC | c.609G>A | p.Trp203* | 42(25.77) |
| c.658_660delAAG | p.Lys220del | 34(20.86) | |
| c.482G>A | p.Arg161Gln | 22(13.50) | |
| c.80A>G | p.Gln27Arg | 14(8.59) | |
| c.565C>T | p.Arg189Cys | 10(6.13) | |
| c.567dupT | p.Ile190Tyrfs*13 | 7(4.29) | |
| c.365A>T | p.His122Leu | 4(2.45) | |
| c.728delC | p.Pro243Leufs*48 | 3(1.84) | |
| c.452A>G | p.His151Arg | 3(1.84) | |
| c.271dupA | p.Arg91lysfs*14 | 3(1.84) | |
| c.688C>T | p.Arg230* | 3(1.84) | |
| c.481C>T | p.Arg161* | 2(1.23) | |
| c.315C>G | p.Tyr105* | 2(1.23) | |
| c.81+1G>A | / | 2(1.23) | |
| c.617G>A | p.Arg206Gln | 2(1.23) | |
| c.1A>G | p.0? | 2(1.23) | |
| c.394C>T | p.Arg132* | 2(1.23) | |
| c.276+1G>A | / | 1(0.61) | |
| c.388T>C | p.Tyr130His | 1(0.61) | |
| c.445_446delTG | p.Cys149Hisfs*32 | 1(0.61) | |
| c.57_58insT | p.Gly20Trpfs*14 | 1(0.61) | |
| c.616C>T | p.Arg206Trp | 1(0.61) | |
| c.328_331delAACC | p.Asn110Aspfs*13 | 1(0.61) | |
| MMUT | c.1663G>A | p.Ala555Thr | 12(15.38) |
| c.1208G>A | p.Arg403Gln | 12(15.38) | |
| c.729_730insTT | p.Asp244Leufs*39 | 9(11.54) | |
| c.1106G>A | p.Arg369His | 8(10.26) | |
| c.914T>C | p.Leu305Ser | 6(7.69) | |
| c.1677-1G>A | / | 3(3.85) | |
| c.323G>A | p.Arg108His | 3(3.85) | |
| c.2179C>T | p.Arg727* | 3(3.85) | |
| c.1630_1631delGGinsTA | p.Gly544* | 2(2.56) | |
| c.626_627insC | p.Lys210* | 1(1.28) | |
| c.1880A>G | p.His627Arg | 1(1.28) | |
| c.2080C>T | p.Arg694Trp | 1(1.28) | |
| c.1280G>A | p.Gly427Asp | 1(1.28) | |
| c.103C>T | p.Gln35* | 1(1.28) | |
| c.652C>T | p.Gln218* | 1(1.28) | |
| c.1630G>T | p.Gly544* | 1(1.28) | |
| c.693C>G | p.Tyr231* | 1(1.28) | |
| c.544_545insA | p.Met182Asnfs*29 | 1(1.28) | |
| c.1105C>T | p.Arg369Cys | 1(1.28) | |
| c.912-1G>A | / | 1(1.28) | |
| c.1809-1G>A | / | 1(1.28) | |
| c.920_923delTCTT | p.Phe307Serfs*6 | 1(1.28) | |
| c.682C>T | p.Arg228* | 1(1.28) | |
| c.970G>A | p.Ala324Thr | 1(1.28) | |
| c.278G>A | p.Arg93His | 1(1.28) | |
| c.755dupA | p.His252Glnfs*6 | 1(1.28) | |
| c.1102delG | p.Val368Serfs*5 | 1(1.28) | |
| c.1560+1G>A | / | 1(1.28) | |
| c.975_976delTAinsGCTCTCTTT | p.Arg326Leufs*17 | 1(1.28) | |
| MMAA | c.988C>T | p.Arg330* | 2(40.00) |
| c.1076G>A | p.Arg359Gln | 2(40.00) | |
| c.650T>A | p.Leu217* | 1(20.00) | |
| MMAB | c.584G>A | p.Arg195His | 1(20.00) |
| c.571C>T | p.Arg191Trp | 1(20.00) | |
| c.569G>A | p.Arg190His | 1(20.00) | |
| c.572G>A | p.Arg191Gln | 1(20.00) | |
| c.582_583insGA | p.Arg195Aspfs*20 | 1(20.00) |
注:/:内含子变异,该变异不影响氨基酸改变,仅影响剪切 /:intron variation,which does not affect amino acid changes,but only affects splicing
2 000例新生儿通过该技术对MMA相关的8个基因(MMACHC、MMUT、MMAA、MMAB、MMADHC、LMBRD1、ABCD4、HCFC1)174个基因位点进行筛查。共检测到3个基因的致病变异,其中MMACHC基因检测到11种致病突变,MMUT基因检测到10种致病突变,MMAB基因检测到1种致病突变。基因筛查确诊MMA患儿1例,为c.481C>T/c.80A>G的复合杂合变异。该患儿新生儿筛查结果C3、C3/C2升高,召回复查生化结果符合合并型MMA诊断。未确诊新生儿共67例携带MMA相关致病基因,携带率为3.35%,其中47例携带MMACHC基因,携带率为2.35%,19例携带MMUT基因,携带率为0.95%,1例携带MMAB基因,携带率为0.05%。MMACHC基因的热点突变为c.609G>A(38.30%)、c.658_660delAAG(14.89%)、c.482G>A(14.89%)、c.80A>G(8.51%);MMUT基因的热点突变为c.729_730insTT(26.32%)、c.1663G>A(15.79%)、c.323G>A(15.79%)、c.424A>G(10.53%)。各筛查位点的携带情况见表3。
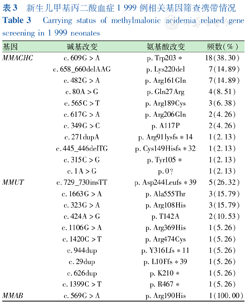
新生儿甲基丙二酸血症1 999例相关基因筛查携带情况
Carrying status of methylmalonic acidemia related gene screening in 1 999 neonates
新生儿甲基丙二酸血症1 999例相关基因筛查携带情况
Carrying status of methylmalonic acidemia related gene screening in 1 999 neonates
| 基因 | 碱基改变 | 氨基酸改变 | 频数(%) |
|---|---|---|---|
| MMACHC | c.609G>A | p.Trp203* | 18(38.30) |
| c.658_660delAAG | p.Lys220del | 7(14.89) | |
| c.482G>A | p.Arg161Gln | 7(14.89) | |
| c.80A>G | p.Gln27Arg | 4(8.51) | |
| c.565C>T | p.Arg189Cys | 3(6.38) | |
| c.617G>A | p.Arg206Gln | 2(4.26) | |
| c.349G>C | p.A117P | 2(4.26) | |
| c.271dupA | p.Arg91lysfs*14 | 1(2.13) | |
| c.445_446delTG | p.Cys149Hisfs*32 | 1(2.13) | |
| c.315C>G | p.Tyr105* | 1(2.13) | |
| c.1A>G | p.0? | 1(2.13) | |
| MMUT | c.729_730insTT | p.Asp244Leufs*39 | 5(26.32) |
| c.1663G>A | p.Ala555Thr | 3(15.79) | |
| c.323G>A | p.Arg108His | 3(15.79) | |
| c.424A>G | p.T142A | 2(10.53) | |
| c.1106G>A | p.Arg369His | 1(5.26) | |
| c.1420C>T | p.Arg474Cys | 1(5.26) | |
| c.944dup | p.Y316Lfs*11 | 1(5.26) | |
| c.29dup | p.L10Ffs*39 | 1(5.26) | |
| c.626dup | p.K210* | 1(5.26) | |
| c.1399C>T | p.R467* | 1(5.26) | |
| MMAB | c.569G>A | p.Arg190His | 1(100.00) |
MMA是目前报道发病率最高的有机酸代谢病,临床表现缺乏特异性,可导致多个系统损伤,严重时可危及生命,以神经系统损伤最为常见,如未能及时治疗可留有严重的后遗症,早期规范的治疗能够降低病死率,改善预后[2,7,8]。本研究中2种基因筛查检测新生儿MMA致病基因的携带率均高于青岛地区筛查的18个热点突变的携带率(2.5%)[9]。MMA在世界各地的患病率差异较大,据报道,意大利MMA的患病率约为1∶22 727[10],美国约为1∶158 730[11],日本约为1∶120 000[12]。相比之下我国的发病率更高,但也存在明显的地域差异,呈南低北高的趋势,我国台湾MMA的患病率约为1∶101 625[13],上海新华医院根据2003年至2008年新筛情况得出MMA的患病率为1∶28 000[14],新乡MMA的患病率约为1∶6 264[15]。根据已有报道[6],山东地区的MMA在全国的发病率最高。本研究对济南市11年36万余例新生儿筛查MMA得出济南地区MMA的患病率为1∶3 567,较之前的MMA患病率报道[6]更高。目前的生化筛查方法通过串联质谱技术筛查MMA是非常成功且高效的一线筛查方法。
在筛查及复查均怀疑MMA的新生儿中有82例完成了基因检测,其中76例获得基因确诊,2例仅查到1个基因突变,4例未检测到MMA相关基因变异。未获得基因确诊的6例早期筛查指标C3/C2、C3及尿甲基丙二酸均轻度升高,予维生素B12治疗后代谢指标恢复正常,因基因检测结果不支持MMA诊断,结合患儿生化指标异常程度较轻,予停药随访,监测代谢指标正常,排除诊断。因此,基因检测除了明确MMA患儿的基因突变类型,还可对生化指标轻度异常的情况进行早期鉴别。此外MMA基因型与表型之间有着较强的相关性,基因检测对MMA的治疗也有重要的指导意义[2,16] 。
本研究发现c.609G>A是MMACHC基因最常见的变异类型,这与既往研究结果一致[17]。单纯型MMA患儿中mut型是最常见的亚型,本研究中确诊的MMA患儿基因显示,MMUT基因最常见的变异为c.1663G>A(12.50%)及c.1106G>A(12.50%)。4 800例的基因筛查结果显示MMUT基因携带率最高为c.1663G>A(15.38%)和c.1208G>A(15.38%),而另一项2 000例的基因筛查结果示MMUT基因携带率最高为c.729_730insTT(26.32%)和c.1663G>A(15.79%)。综合分析c.1663G>A、c.1106G>A、c.1208G>A和c.729_730insTT均为济南地区MMUT基因的热点变异。一项关于中国26个省市314例MMA患者的研究发现,c.729_730insTT是MMUT基因最常见变异[2];而另一项甘肃地区的研究则认为c.323G>A、c.917C>T及c.984delC为MMUT最常见的变异。这些研究结果的差异可能与样本量小有关,也可能与地域不同有关。
根据新生儿基因筛查发现的MMA相关基因携带率推测MMA的患病率可能高于目前根据生化筛查确诊患儿计算的MMA患病率,这可能与新生儿基因筛查样本量相对较小有关,但因MMA患儿存在一部分晚发型患儿,在新生儿早期代谢指标还未发生异常时检测不出,导致目前的新生儿生化筛查可能漏筛部分MMA晚发型患儿,所以新生儿基因筛查可作为新生儿疾病筛查的重要补充。目前关于大样本MMA生化筛查与基因筛查的对比研究较少,Luo等[18]对1 127例接受过新生儿疾病生化筛查的个体进行了基因筛查,筛查出4例葡萄糖-6-磷酸脱氢酶患儿,1例原发性肉碱缺乏症患儿,该患儿生化筛查结果为阴性。敖桢桢等[19]报道了广东地区2万例新生儿完成串联质谱筛查后对质谱检测阳性、疑似及临床高度怀疑遗传代谢病的109例患儿进行基因测序分析,结果提示串联质谱分析存在一定的假阳性率和假阴性率,串联质谱法联合高通量测序分析可有效降低串联质谱筛查的假阳性率和假阴性率,并可尽早明确患者致病基因,为后续的临床治疗和遗传咨询提供依据。新生儿基因筛查周期一般在2周左右,且与新生儿疾病筛查同步进行,与以往的筛查异常后再进行基因检测相比缩短了MMA患儿的确诊时间。进行基因筛查时,对基因的检测方式有2种,一是对全基因进行筛查,另一种是仅对热点致病基因进行筛查。全基因筛查的检测范围广,易发现新发变异,但检测所需血量较大且对于意义不明的变异结果解读困难,临床应用存在较大挑战,热点基因筛查所需检测血量较少且临床应用相对简单,是目前应用较多的方案,但该方法无法发现新发变异。未来新生儿基因筛查技术将会逐渐在临床推广,但选择哪种检测方案还有待进一步探讨。
随着串联质谱技术的普及,通过新生儿筛查确诊的MMA患儿越来越多,但对于严重的早发型患儿,患儿可能在胎儿时期已发生损伤,待新生儿筛查确诊时可能已对患儿造成不可逆损伤,本研究提供了群体新生儿基因筛查得出的人群携带MMA相关致病基因的热点突变,可建议本地域育龄夫妇进行MMA相关致病基因的携带者筛查,当夫妻同为MMA相关基因携带者时建议通过遗传咨询避免MMA患儿的出生,将MMA防控关口前移到二级甚至一级预防措施。
所有作者均声明不存在利益冲突

























