
脊髓性肌萎缩症(SMA)是一种严重的遗传性神经肌肉病,是由于运动神经元存活基因1(SMN1)致病性变异导致的编码产物SMN蛋白缺乏所致。根据患者起病年龄及获得的最大运动功能,SMA分为4个亚型,其中最常见且表型最严重的为SMA1型。自然病程中,大多数SMA1型患儿2岁内死于呼吸衰竭,2~4型患者均有不同程度缓慢进展的肌无力和肌萎缩。疾病修正治疗问世以前,以康复训练、呼吸及营养支持为主的综合治疗是延缓SMA患者病程进展、提高生存率的唯一手段,但由于未从根本上解决SMN蛋白的缺乏,因此患者无法取得运动里程碑的改善。5年来,先后有3种药物获批用于治疗SMA。越来越多的临床试验及真实世界研究数据表明,疾病修正治疗药物可以有效维持各型SMA患者的运动功能,且可促进部分患者获得里程碑进步,显著降低死亡率。但是,不同亚型、病情和病程的患者对药物治疗的反应可能存在明显差异,因此,选择合适的治疗方案,保证患者生活质量尤为重要。本综述主要讨论SMA治疗方面的最新进展及面临的新挑战。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
脊髓性肌萎缩症(spinal muscular atrophy,SMA)是由于运动神经元存活基因1(SMN1;OMIM 600354)双等位基因致病性变异所致,近期有研究报道中国新生儿中发病率为1/9 788,人群携带率高达1/50[1,2],是导致2岁以下婴幼儿死亡最常见的遗传病。由于SMN1基因缺失,SMN蛋白表达显著减少,从而导致脊髓前角运动神经元变性,引起进行性肌无力及萎缩。自1995年法国Lefebvre等[3]发现SMN1基因的致病性变异是SMA的主要致病原因后,人们又进一步对SMA的病理生理过程进行研究,尽管SMN蛋白缺乏引起SMA的机制尚不完全明确,但旨在提高SMN蛋白水平的药物已相继问世。2016年12月Nusinersen(诺西那生)的获批,标志着SMA进入了疾病修正治疗(disease-modifying therapy, DMT)时代,其后5年里,共3种药物获批治疗SMA。临床试验及真实世界研究结果表明,这些药物可以改善SMA所有亚型的运动功能,不同程度提高患者的生活质量[4,5,6,7]。本综述总结了SMA的分子机制、临床表现及目前的治疗进展,旨在为SMA的临床诊疗提供参考,还进一步讨论了该领域可能面临的新挑战。
SMN1基因定位于染色体5q11.2-q13.3,SMN2基因是SMN1基因的同源基因,两者功能差异主要由外显子7中一个碱基(c.840 C>T)的不同所造成,该变异导致SMN2基因外显子7被剪切,仅产生10%~15%有功能的全长SMN蛋白,大部分翻译出无功能截短SMN蛋白。在人群中SMN2基因的拷贝数为0~8,大多数人存在1~4个拷贝。在SMA患者中,SMN2基因拷贝数与疾病严重程度呈负相关,即SMN2拷贝数越多,表型越轻[8]。
目前尚不明确SMN蛋白缺乏如何引起SMA,但SMN蛋白的许多生物学功能已被阐明。SMN蛋白在所有组织中广泛表达,参与调节RNA代谢的各个方面,其与8种蛋白(Gemin 2~8和Unrip)形成复合物,不仅在核内小核糖核蛋白颗粒、小核仁核蛋白复合物、富含尿苷7的核内小核糖核蛋白(U7 snRNP)等核糖核蛋白的生物发生中发挥作用,还参与了转录后mRNA加工和转运、翻译调节、DNA重组和修复等过程[9]。SMN蛋白也广泛存在于运动神经元的轴突中,参与了重要的神经元生理活动,如神经元细胞骨架维持及其动力学、信号转导、凋亡、内吞、自噬等,从而起到促进轴突生长、维持神经元稳态的作用[10]。
根据患者起病年龄及可达到的最大运动功能,SMA通常被分为4个亚型,分别为SMA1型、SMA2型、SMA3型和SMA4型。SMA1型为最严重的表型,该型患者在出生6个月内起病,不能达到独坐里程碑,随疾病进展最终出现呼吸和吞咽功能下降,在没有药物治疗及呼吸支持的情况下,大多数1型患者2岁内死于呼吸衰竭或其他严重并发症。SMA2型通常起病时间为6~18个月,进展较1型慢,可达到独坐里程碑,但不能独走,多数SMA2型患者可活到成年期。SMA3型及4型临床表现相对较轻,患者均可达到独走,肌无力症状通常随年龄增长而加重,部分SMA3型患者最终丧失独走能力[11]。也有学者根据患者起病年龄进一步细化分型,如1型分为1a(即0型,宫内发病)、1b和1c型,2型分为2a、2b型,3型分为3a和3b型[12]。越来越多的证据表明,严重类型的SMA患者可以表现出神经肌肉系统以外的症状,如心血管系统(先天性心脏病、心律失常、心肌纤维化等)[13]、消化系统(便秘、腹胀、胃食管反流、脂肪肝等)、代谢紊乱(血脂异常、线粒体功能障碍、低血糖、高血糖、胰岛素抵抗、酮症等)等[14]。
随着DMT的开展,SMA患者的表型谱发生了变化,因此,原有表型分类方法将有所改变[12]。
DMT是指通过影响疾病的病理生理过程,对该疾病的病程产生有益结局的治疗或干预手段[15]。在过去几年中,SMA的DMT取得了突破性进展,治疗研究主要围绕增加SMN蛋白水平及增加肌肉力量(表1),其他治疗靶点如稳定SMN蛋白、神经元保护、干细胞治疗等也在动物模型中取得了一定的效果,但仍在临床前研究阶段。
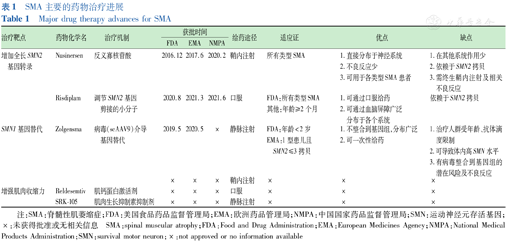
SMA主要的药物治疗进展
Major drug therapy advances for SMA
SMA主要的药物治疗进展
Major drug therapy advances for SMA
| 治疗靶点 | 药物化学名 | 治疗机制 | 获批时间 | 给药途径 | 适应证 | 优点 | 缺点 | ||
|---|---|---|---|---|---|---|---|---|---|
| FDA | EMA | NMPA | |||||||
| 增加全长SMN2基因转录 | Nusinersen | 反义寡核苷酸 | 2016.12 | 2017.6 | 2020.2 | 鞘内注射 | 所有类型SMA | 1.直接分布于神经系统2.不良反应少3.可用于各类型SMA患者 | 1.在其他系统作用少2.依赖于SMN2拷贝3.需终生鞘内注射及相关不良反应 |
| Risdiplam | 调节SMN2基因剪接的小分子 | 2020.8 | 2021.3 | 2021.6 | 口服 | FDA:所有类型SMA其他:年龄≥2个月 | 1.可通过口服给药2.可通过血脑屏障广泛分布于各个系统 | 依赖于SMN2拷贝 | |
| SMN1基因替代 | Zolgensma | 病毒(scAAV9)介导基因替代 | 2019.5 | 2020.5 | × | 静脉注射 | FDA:年龄<2岁 EMA:1型患儿且SMN2≤3拷贝 | 1.不整合到基因组,分布广泛2.可一次性给药 | 1.治疗人群受年龄、抗体滴度限制2.可导致体内高SMN水平3.有病毒整合到基因组的潜在风险及不良反应 |
| × | × | × | 鞘内注射 | × | × | × | |||
| 增强肌肉收缩力 | Reldesemtiv | 肌钙蛋白激活剂 | × | × | × | 口服 | × | × | × |
| SRK-105 | 肌肉生长抑制素抑制剂 | × | × | × | 静脉注射 | × | × | × | |
注:SMA:脊髓性肌萎缩症;FDA:美国食品药品监督管理局;EMA:欧洲药品管理局;NMPA:中国国家药品监督管理局;SMN:运动神经元存活基因;×:未获得批准或无相关信息 SMA:spinal muscular atrophy;FDA:Food and Drug Administration;EMA:European Medicines Agency;NMPA:National Medical Products Administration;SMN:survival motor neuron;×:not approved or no information available
Nusinersen是SMN2基因的剪切修饰剂,是含18个碱基的反义寡核苷酸(ASO),其通过与SMN2基因上反向调控外显子7表达的序列相结合,抑制剪接因子的作用,使外显子7在剪接过程中被保留,能够产生全长mRNA,翻译出更多全长SMN蛋白。2011年就已在动物试验中证实,通过鞘内注射Nusinersen,可以显著增加SMA小鼠脊髓前角运动神经元中SMN蛋白水平,从而改善运动功能和生存率[16]。随后在多项临床试验中,Nusinersen均显示出较好的安全性及疗效,部分患儿实现了超越其自然发育里程碑的进步,绝大多数患儿显示有意义的运动功能评分改善。在一项2期开放标签研究(NURTURE,NCT02386553)中发现,症状前开始应用Nusinersen治疗的患儿,可能实现正常发育里程碑[17]。Nusinersen目前已被全球包括中国在内的多个国家批准用于婴儿、儿童和成人SMA治疗,适用于全部类型SMA患者。由于无法通过血脑屏障且有一定半衰期,Nusinersen需要反复通过鞘内注射给药,每次给药剂量为12 mg (5 mL),分别在第0、14、28和63天予4次负荷剂量,之后每4个月予1次维持剂量。对于接受脊柱侧弯矫形术(如脊柱后路融合术)的患儿,须预留Nusinersen鞘内注射通道,对于合并脊柱侧弯或脊柱后路融合术后患者,可于B超或CT引导下行腰椎穿刺[18]。不同国家及地区接受Nusinersen治疗的真实世界数据表明,Nusinersen治疗对各型患者均有运动功能改善作用[6,19,20]。Nusinersen的高剂量临床试验正在进行中,包括一项2/3期临床研究(DEVOTE,NCT04089566)和一项3b期临床研究(ASCEND,NCT05067790)等。Nusinersen的不良反应主要为腰椎穿刺术相关并发症,如头疼、背痛、呕吐等,药物上市后有报道治疗后发生交通性脑积水[21] 。此外,在其他ASO的临床试验中曾报道过严重血小板减少症,其中1例出现了致死性颅内出血[22]。在迟发型SMA患者中进行的3期研究中(CHERISH,NCT02292537),有2例治疗组患儿曾出现显著血小板减少,其中1例患儿出现在Nusinersen治疗的第28、273天[23]。以上研究中均无关于血小板减少引起出血性事件的报道。在其他ASO中也有关于肾毒性的报道,目前推测可能与反复给药导致的药物累积相关[24]。在Nusinersen的临床研究中,仅少数患儿在治疗后出现尿蛋白、尿素氮及血清肌酐轻度升高,在真实世界研究中未出现关于Nusinersen肾毒性的报道。ASO引起血小板减少、肾毒性的潜在机制仍在进一步研究中。尽管目前尚未证实Nusinersen临床研究中的血小板减少及肾功能相关指标异常与药物相关,但基于既往ASO异常的实验室数据,仍建议在基线及每次给药前均完善血小板、尿蛋白、肾功能等检测。目前一项旨在观察Nusinersen上市后安全性的研究(NCT04419233)正在中国进行,计划结束时间为2023年。
Risdiplam作为目前唯一口服小分子药物,在被批准前就受到广泛关注。其通过选择性与SMN2基因前mRNA中的2个剪接调控位点结合,增强U1 snRNP的识别能力,同样改变剪切,从而增加全长SMN mRNA和蛋白水平[25]。Risdiplam可透过血脑屏障,在全身组织中广泛分布。Risdiplam目前已在包括中国在内的多个国家批准用于年龄≥2个月的患者,并于2022年6月获美国食品药品监督管理局(FDA)批准治疗2个月以下的SMA患儿。其可以通过口服或鼻饲给药,每日1次,药物剂量取决于年龄及体重:<2岁的患儿推荐剂量为0.20 mg/kg,≥2岁且体重<20 kg推荐剂量0.25 mg/kg,≥2岁且体重≥20 kg的患儿推荐剂量5.00 mg,在分发给患者前须由医疗卫生专业人员配制成口服溶液。FIREFISH是一项在SMA1型患儿中开展的开放标签、多中心2/3期临床研究,中国有2个中心参与了其第2部分疗效及安全性研究。2年的研究数据显示,61%的患儿可达到无支撑独坐至少5 s,44%的患儿可达到无支撑独坐至少30 s,76%的患者费城儿童医院婴儿神经肌肉疾病测试得分≥40分,83%的患儿可无需永久性通气生存[26]。在2型或3型SMA患者中同样可以显著改善运动功能,SUNFISH研究第二部分1年的数据发现Risdiplam组与安慰剂组之间的运动功能32项评估量表总评分相较于基线的变化差异有统计学意义[27]。另一项研究RAINBOWFISH于2022年3月公布的中期数据显示,所有症状前患儿(共6例)在接受至少12个月的Risdiplam治疗后,均实现了无支撑坐、翻滚和爬行,4例患儿实现了独站,3例患儿实现了独走,患儿均可无需永久性通气生存[28]。在多个Risdiplam临床试验中均未发现严重的不良反应。最常见的不良反应包括发热、皮疹、腹泻、呕吐、便秘等。此外,在Risdiplam的一项临床前研究中发现了与药物剂量相关的视网膜毒性,可能与其在色素视网膜细胞中的累积相关(Risdiplam具有较高的黑色素结合能力)[25]。因此,在Risdiplam临床研究中也进行了长期的眼科评估,但在所有评估中均未发现治疗相关的视网膜毒性,故在其药品说明中未推荐将眼科检查作为用药常规监测内容。关于Risdiplam长期使用的眼科安全性研究目前仍在进行中。
Zolgensma是SMN1基因替代治疗药物,通过自我互补的腺相关病毒血清型-9载体,将1个或多个SMN1基因拷贝传递到运动神经元的细胞核,导入基因可持续表达,但不整合至宿主基因组[29]。Zolgensma是全球第2个被FDA批准用于治疗SMA的药物,尚未在国内应用,目前仅适用于<2岁且腺相关病毒抗体滴度<50 mg/mL的患儿。其优点在于单次静脉注射,无需长期给药,推荐剂量为每千克体重1.1×1014载体基因组。目前该药的多项临床研究尚在进行中,其中SPR1NT研究结果显示,SMN2基因为2拷贝的症状前患者(共14例)在接受Zolgensma治疗后实现了100%生存(未使用呼吸或营养支持),大部分患者(11/14)实现了正常发育里程碑,并在其扩展研究中依旧保持较好的运动功能[30]。该药鞘注制剂的部分临床研究(STRONG研究的高剂量组)因鞘注给药后导致动物背根神经节损伤,于2019年10月被叫停,但在已进行的试验(STRONG研究的低、中剂量组)中,SMA3型患者显示出了明显的改善。近期鞘注制剂的一项3期临床研究(STEER研究)已重新获FDA批准启动。另外,国内自主研发的基因治疗研究目前也在进行中。Zolgensma已报道的不良反应包括发热、肝毒性、血小板减少、血栓性微血管病等,其中肝毒性最常见。在Zolgensma多个临床试验中均监测到了转氨酶的升高,通常在治疗1周至1个月出现[5],上市后的真实世界研究陆续报道了黄疸、凝血功能异常、急性肝衰竭等肝病表现[31]。由于泼尼松龙可减轻Zolgensma的肝毒性,目前推荐在治疗开始前1 d给予全身皮质类固醇激素治疗。临床试验数据表明,部分患儿在Zolgensma静脉应用后的2个月内可能出现血小板减少[5],但目前机制尚不清楚。上市后的安全性研究也有较多血小板减少的报道,其中19例合并出血性事件,但在临床干预后均能恢复;血栓性微血管病是上市后监测中新发现的不良反应,在已报道的病例中,其发生在给药后的1~2周,临床表现包括呕吐、肾损伤、溶血性贫血等[31]。此外,在临床前研究中曾观察到小鼠的心脏炎症和心内血栓形成,但在临床研究及上市后研究中未报道过明确的心脏毒性,仅在少数病例中观察到心动过缓、肌钙蛋白升高等[5]。Zolgensma长期随访分析(START研究)已于2020年6月11日启动,旨在进一步探索基因治疗的长期疗效及安全性。基于以上可能出现的不良反应,用药前评估及用药后密切的安全性监测是必要的,专家建议在治疗前后对肝功能、血小板计数和肌钙蛋白I等监测至少3个月。
最新研究发现,疾病持续时间、治疗开始时的年龄以及基线功能被确定为潜在的生存相关治疗改变因素,因此强调早期DMT至关重要[32] 。
Reldesemtiv是一种小分子快速骨骼肌肌钙蛋白激活剂,可以通过增加肌钙蛋白C对钙的亲和力,从而增强肌肉的收缩力。可通过口服或鼻饲给药。2期临床试验显示该药对≥12岁的2/3型SMA患者运功和呼吸功能均有改善趋势。
SRK-015是一种单克隆抗体,通过选择性抑制肌生成抑制素,促进肌细胞生长分化,从而改善肌肉力量。可以通过静脉给药。目前进行的TOPAZ 2期临床试验中(NCT03921528),已在2~21岁SMA2型和3型患者中观察到了运动功能的进步。
SMA已进入DMT时代,尽管治疗靶点不尽相同,目的均为增加全长SMN蛋白水平。但是,DMT并非治愈疾病,即使是SMN1基因替代,其治疗后患者病理生理变化仍需要持续关注。临床医师面临很多新的压力:(1)患者及其家庭面临的压力,包括正确知识的获得,治疗的选择,照护困难,终生用药、长期监测,担心患儿生命和个人发展,家庭资源困境,心理社会问题等;(2)专业人员不足:规范化诊治有待进一步提高,尤其是康复训练、营养和呼吸支持、家庭照护指导、围术期管理等;(3)药物和康复治疗辅具等相关治疗保障的压力也不容忽视。由于DMT价格昂贵,目前无法惠及每一个SMA患者,规范化多学科管理仍是大多数患者主要的治疗手段。SMA的多学科管理不应被药物治疗取代,相反应随着药物治疗的发展而加强和完善。总之,临床医师不仅希望延长患者生命的"长度",更希望能拓宽他们生命的"宽度",只有通过不断地思考和创新,才能为更多患者带来希望。
所有作者均声明不存在利益冲突

























