
探讨单核苷酸多态性微阵列(single nucleotide polymorphisms array,SNP array)在染色体异常诊断方面的价值。
应用SNP array和实时定量基因扩增荧光检测系统(qPCR)对一对智力低下的父子的全基因组DNA进行高分辨率分析。分析此家系两位患者临床表现型与染色体异常的相关性,并确定患儿染色体异常片段的来源。
患儿芯片核型为arr[hg19]7q11.23(72722981-74138121)×1;患儿父亲芯片核型为arr[hg19]7q11.23(72722981-74138121)×1。患儿从父亲那里继承了异常的7号染色体,该异常与患者临床表现密切相关。
7号染色体发生的约1.42 Mb的缺失导致这对父子的临床表现为威廉氏症候群(Williams-Beuren Syndrome,WBS)。通过高分辨率的SNP array技术明确了患儿异常片段的来源,并且提供了详细、准确的染色体信息,有助于明确临床症状与患者基因异常的相关性,同时评估了染色体异常的再发风险。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
世界范围内人口生育率逐年下降[1],全球半数左右的国家将面临婴儿出生数量无法维持该国人口规模的困境。作为一项国家战略资源和生产力要素,人口发展情况反映了国情、国力。调查显示[2],我国已婚育龄妇女不孕症的发生率从上世纪80年代的2%~5%,上升到近年的10%左右[3]。
威廉氏症候群(Williams-Beuren syndrome,WBS)是一种遗传性涉及多个系统的疾病,新生儿的发病率为1/20 000~1/7500,主要特征为先天性心脏病(以主动脉瓣狭窄为主)、智力障碍、轻微的身材矮小、面部先天性畸形、泌尿生殖系统畸形以及骨骼异常等。WBS患者有典型的面部特征:高额头、眉间距宽、眼眶骨饱满、鼻梁低、腮骨发育不全、厚嘴唇和长人中。临床诊断通常在儿童时期进行,此时典型的面部变化和认知轮廓变得更加明显。可以使用荧光原位杂交(FISH)或进行重连接依赖式探针扩增(MLPA),或进行微阵列检测(SNP-array/array CGH)以明确染色体异常。WBS患者中90%患者染色体7q11.23处发生1.55 Mb的微缺失,8%患者发生1.84 Mb的微缺失,这些为典型WBS。仅2%患者的缺失或大于1.84 Mb或小于1.55 Mb,被定义为非典型WBS[1]。本文旨在描述1个非典型WBS家系的临床特点及遗传资料,以阐明基因型-表现型的相关性。并评估该家系遗传结果的临床意义,为该病的临床筛查和遗传咨询提供更多信息。
患儿,男,6岁,因智力低下查因到铁道警察学院刑事科学技术系理化物证检验研究中心就诊。患儿身高正常(1.10 m),多动,注意力短暂,喜欢交流,无陌生感,但语言无逻辑,口齿不清。患儿嘴唇较厚、外翻,颧骨低,鼻梁低,牙齿稀疏。患儿父亲,男,37岁,因自身智力稍低,有智力低下患儿生育史,其妻智力正常,夫妻双方为生育健康孩子就诊。患儿父亲个头较矮(1.50 m),嘴唇较厚、外翻,颧骨低,鼻梁低,牙齿稀疏。智力稍低,在家人的多番努力下初中毕业。幼时因主动脉瓣狭窄做过心脏手术。
用TIANamp Micro DNA (Cat.#DP316)(北京天根生化科技有限公司)提取全基因组DNA。用超微量分光光度计(Nanovue Plus,英国GE Healthcare)测定DNA浓度,之后冻存于-20 ℃备用。
按照常规方法处理标本,制备染色体,进行G显带。采用GSL120染色体自动扫描系统(德国Leica)采集核型,应用Cytoversion software(德国Leica)分析核型,按照人类细胞遗传学国际命名体制(ISCN-2009)标准描述核型。
用Human CytoSNP-12 DNA Analysis BeadChip(美国Illumina公司)进行芯片的杂交和扫描,扫描结果用GenomeStudio software进行分析。
7q11.23缺失处基因拷贝数检测引物:primer1正向引物5'-CTTCAAGGAGCCCCTGATGC-3';60℃;primer1反向引物5'-ACAGATGAGCTCCTGGTCCT-3',60℃,259 bp;primer2正向引物5'-TGACAGGCTGTTTA AGGCGG-3',60 ℃;primer2反向引物5'-GATTTCCAGGTAGCTGGGGG-3',60 ℃,194 bp。
芯片扫描结果得到的异常片段通过查阅DECIPHER、OMIM、NCBI、GeneCard等数据库进行异常片段致病性分析。
结果如图1所示,在本技术分辨率范围内,该家系患者的染色体未发现可疑异常。
SNP array结果显示患儿及其父亲在7q11.23存在约1.42 Mb(72722981~74138121)的缺失,他们的芯片核型为arr[hg19]7q11.23(72722981~ 74138121)×1。该异常片段包含24个编码蛋白的基因和2个microRNAs:TRIM50、FKBP6、FZD9、BAZ1B、BCL7B、TBL2、MLXIPL、VPS37D、DNAJC30、WBSCR22、STX1A、ABHD11、CLDN3、CLDN4、WBSCR27、WBSCR28、ELN、LIMK1、EIF4H、RFC2、LAT2、CLIP2、GIF2IRD1、GTF2I和MIR4284、MIR590(图2)。
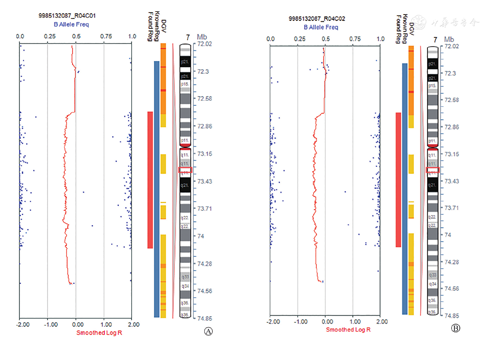
A示父亲del(7) q11.23 1.42 Mb;B示儿子del(7) q11.23 1.42 Mb
为了进一步验证SNP array的结果,利用qPCR检测7q11.23缺失处基因拷贝数变化。选取靠近缺失边缘的TRIM50和GTF2I两个基因设计qPCR引物(primer1&2),检测结果显示当正常样本的相对拷贝数为1时,患者父子TRIM50基因的相对拷贝数分别为0.47和0.45,GTF2I基因的相对拷贝数分别为0.52和0.49。说明患者及其儿子7q11.23缺失处基因拷贝数减少。
经数据库分析结合患者的临床症状,我们认为该对父子患者为WBS。WBS患者是由于染色体7q11.23发生1~2 Mb的微缺失造成的,在该段区域包含28个基因。WBS患者中90%患者发生1.55 Mb的微缺失,8%患者发生1.84 Mb的微缺失,这些患者为典型WBS。仅2%患者的缺失或大于1.84 Mb或小于1.55 Mb,被定义为非典型WBS。在7号染色体长臂的着丝粒区(centromeric LCR) 、中间区(medial LCR)和端粒区(telomeric LCR)有3个大片段重复序列(low-copy repeat elements,LCRs),每个区域都包括3个不同的区段,称为"block A"、"block B"和"block C",它们通过非等位基因同源重组(non-allelic homologous recombination,NAHR)构成每个WBS患者的侧翼序列。位于中间区的block B(Bm)包括3个基因,GTF2I、NCF1和GTF2IRD2,它们对应位于着丝粒区的block B(Bc)的3个假基因,GTF2IP1、NCF1P1和GTF2IRD2P1,以及位于端粒区(Bt)的GTF2IP2、NCF1P2和GTF2IRD2P2。大部分WBS患者(90%)发生约1.5 Mb微缺失是由于Bc和Bm区发生NAHR导致的。位于中间区的block A(Am)包括3个假基因,STAG3L2、PMS2L5和GATS-L,它们对应位于着丝粒区的block A (Ac)的1个STAG3L3非编码基因和2个拷贝的非编码基因PMS2L和WBSCR19。相对比较罕见的5%的WBS患者是由于Ac和Am区的LCRs发生NAHR导致的[2,3]。
本研究的2例WBS患者缺失区域包含24个基因。TRIM50编码一种E3泛素连接酶,它与其它泛素交联酶结合形成二聚体或三聚体发挥作用,有研究显示该基因异常与WBS相关[4]。FKBP6编码一种肽基脯氨酸顺反异构酶,参与免疫调节及基本的细胞进程,如:蛋白的折叠和转运。一些WAS患者该基因缺失。还有报道认为该基因异常可能造成男性不育[5],但不是一定造成男性不育[6]。FKBP6单倍计量不足会造成特定的心血管异常和骨骼肌异常[7]。FZD9编码WNT信号通路受体,该基因主要表达于大脑、睾丸、眼、骨骼肌和肾脏[8]。BAZ1B属于溴结构域蛋白成员,是一种非典型的酪氨酸蛋白激酶,作为转录调节因子参与染色质重塑,BAZ1B计量不足很可能是造成WBS患者心血管和骨骼肌异常的重要原因[9]。BCL7B在心脏、骨骼肌和胎儿肝脏中高表达,WBS患者中该基因常发生缺失[10]。TBL2基因编码β-转导样蛋白2,该基因异常会造成WBS病人心血管和骨骼肌异常[11]。MLXIPL编码一个基本的螺旋-环-螺旋亮氨酸拉链转录因子,属于Myc/Max/Mad超家族成员,它们形成异源二聚体发挥作用,该基因异常与肝细胞癌和WBS等疾病相关[12,13]。VPS37D编码小泡分选相关蛋白,参与小泡运输,分选内吞的泛素化的小体进入不同的小泡中,可能参与细胞的生长和分化,WBS患者常发生缺失[14]。DNAJC30属于分子伴侣超家族成员,功能未见报道。WBSCR22编码的蛋白包括核定位信号和S-腺苷-L-甲硫氨酸模序,具有DNA甲基化作用,可能参与苯丙氨酸和组氨酸代谢相关信号通路[15,16]。STX1A编码突触融合蛋白,是神经系统特异蛋白,位于突触前膜的突触囊泡里,是重要的离子通道调节因子和促进突触囊泡释放,可能介导精子顶体反应[17]。ABHD11编码的蛋白具有水解酶活性,具体生物学功能报道较少。CLDN3和CLDN4编码紧密连接蛋白,作为一道物理屏障阻止水和溶质自由穿过细胞间隙,该基因异常会导致胰母细胞瘤和粘液表皮样癌等多种实体瘤[18]。WBSCR27和WBSCR28属于甲基转移酶超家族成员,涉及WBS患者心血管和骨骼肌异常。ELN是弹性纤维的重要成分,富含疏水性氨基酸,该基因的缺失和突变会造成主动脉瓣狭窄和皮肤松弛症[19]。LIMK1编码LIM结构域蛋白激酶,属于丝氨酸/苏氨酸蛋白激酶,调节激动蛋白的极化从而影响细胞骨架。同时还能刺激轴突生长,可能调节脑发育。该基因缺失在WBS中可能影响患者的视觉空间建构性认知[20]。EIF4H编码真核细胞翻译起始因子,在mRNA水平刺激蛋白翻译[21]。RFC2编码复制因子C,又名激活因子C,主要负责与ATP结合促进细胞的存活,该基因也位于WBS重要区域[22]。LAT2编码T细胞激活连接器,参与肥大细胞中的FCER1信号通路及B细胞BCR信号通路。该基因在脾脏、外周血淋巴细胞和淋巴结生发中心中高表达,在T细胞中无表达[23]。CLIP2编码细胞质连接蛋白,介导特异的模型细胞器和微管间的相互作用,在神经细胞中连接微管到树枝状片状体,控制大脑特异细胞器的位移[24]。GTF2I编码的磷酸蛋白是一种通用转录因子,它结合于一些基因的启动子区,与其它因子(如SRF、PHOX1)等形成复合物,能显著地影响WBSCR27的表达,可能也影响EIF4H的表达,WBS患者的这个基因多发生缺失[25,26]。经典WBS患者为1.5~1.84 Mb缺失,包含26~28个基因,本家系为<1.5 Mb的非经典缺失,包含24个基因。缺失区域中包含几个重要基因:ELN、LIMK1、CLIP2、GTF2IRD1和GTF2I,其中ELN为剂量敏感基因,单倍剂量不足会造成先天性心血管畸形,LIMK1、CLIP2、GTF2IRD1和GTF2I基因异常与WBS患者特有的认知表型、运动功能和颅面特征相关[27]。
综上所述,我们通过高分辨率的SNP array技术详细、准确分析了该对父子的染色体异常片段,为其提供有价值的遗传咨询和评估后代染色体异常的再发风险。患者妻子染色体正常,再次怀孕正常胎儿的可能性为50%,我们建议患者妻子再次妊娠时进行胎儿SNP array产前检测。
所有作者均声明不存在利益冲突

























