
探讨先天性糖基化障碍(congenital disorder of glycosylation,CDG)Ⅱb型的临床特征及遗传学特点,为CDG-Ⅱb型患儿的早期诊断、治疗和预后提供依据。
对南方医科大学附属深圳妇幼保健院新生儿科收治的1例CDG-Ⅱb型患儿资料进行回顾性分析。以"先天性糖基化障碍Ⅱb型"、"先天性糖基化缺陷Ⅱb型"、"甘露糖基-低聚糖葡萄糖苷酶(mannosyl-oligosaccharide glucosidase,MOGS)"和"congenital disorders of glycosylation type Ⅱb"、"congenital defects of glycosylation type Ⅱb、"MOGS"等为关键词,分别对中国知网、维普数据库、万方数据库、生物医学文献数据库(PubMed)、Web of Science数据库自建库至2019年8月收录的文献进行检索,总结CDG-Ⅱb型患儿的临床特征及遗传学特点。
本例患儿女,因"肌张力低下2 d"入院,主要表现为异常面部特征、肌张力低下、低通气、呼吸暂停、需鼻饲喂养、肝大、外生殖器发育不全,全外显子高通量测序分析发现编码MOGS的基因存在2处复合杂合突变,c.1598delT(p.F533Sfs*40)来自母亲,c.1619G>C(p.R540P)来自父亲,均为未报道过的突变。文献检索共收集4篇文献,包括本例共7例CDG-Ⅱb型患儿,其中表现为面部畸形6例;生殖器发育不全、肌张力低下、神经发育迟缓、喂养困难及肝大各5例;听力异常、低通气或呼吸暂停、低丙种球蛋白血症各4例;惊厥、头颅磁共振异常各3例。基因检测均为复合杂合突变,3例存活,4例死亡。
生后有异常面部特征、肌张力低下、惊厥、低通气、呼吸暂停、喂养困难、肝大、外生殖器发育不全、低丙种球蛋白血症等临床表现的患儿,应高度怀疑CDG-Ⅱb型,多数患儿多系统受累,预后不良。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
先天性糖基化障碍(congenital disorder of glycosylation,CDG)是由于蛋白质或脂质糖基化缺陷引起的一组先天性代谢性疾病,常累及多个系统,且多数治疗效果不佳。该病自1980年被发现以来,现已报道100多种不同类型的糖基化障碍性疾病[1,2]。根据CDG糖蛋白形成缺陷发生的环节不同,分为CDG-Ⅰ型和CDG-Ⅱ型,因缺陷酶不同再分为各种亚型[3],其中CDG-Ⅱb型非常罕见,是由编码甘露糖基-低聚糖葡萄糖苷酶(mannosyl-oligosaccharide glucosidase,MOGS)基因缺陷引起的常染色体隐性遗传病。本文就我院收治的1例CDG-Ⅱb型患儿进行报道,并对相关文献进行复习,探讨并总结CDG-Ⅱb型患儿的临床特点及预后。
患儿女,生后2 d,因"肌张力低下2 d"转入我院。患儿系第1胎第1产,胎龄39+2周,因"臀位"剖宫产娩出,出生体重4 000 g,出生史无特殊。父母体健,外貌正常,否认近亲结婚,无家族遗传病史。
入院查体:身长47 cm,头围35 cm,体重3 970 g。神志清,反应欠佳,哭声弱,全身皮肤松弛,前囟已闭,前额狭小,满月脸,头发、眉毛浓密,外耳廓可见密集毛发,睫毛长,眼裂小,眼距宽,心肺查体正常,肝肋下2 cm,脾肋下1 cm,质中,大阴唇发育不全,四肢肌张力低下。见图1。
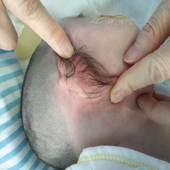
实验室及影像学检查:血常规正常,C反应蛋白21 mg/L,降钙素原1.18 ng/ml,血气分析、肝肾功能及心肌酶谱正常,微小病毒B19阴性,免疫球蛋白、甲状腺功能、促肾上腺皮质激素、皮质醇、甲状旁腺激素正常。尿有机酸及血串联质谱检测正常;微阵列未检测出具有临床意义的微缺失和微重复,染色体正常。头颅磁共振、振幅整合脑电图正常。心脏超声示右心偏大,室间隔增厚,房水平双向分流,动脉导管未闭,肺动脉高压,估测肺动脉压52 mmHg。
患儿住院后诊断新生儿肺炎,予无创呼吸支持、抗感染等治疗。因吸吮、吞咽差,需鼻饲喂养;易出现胃食管反流,体位喂养及药物治疗效果差。经家属同意取患儿及父母外周血行基因检测,基因结果示CDG-Ⅱb型,住院28 d家长放弃治疗签字出院,电话随访患儿出院当天死亡。
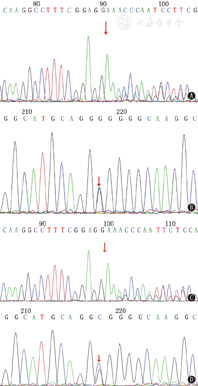
全外显子组高通量测序结果回报,患儿MOGS基因存在复合杂合突变。见图2。患儿外显子4存在c.1598delT移码突变,来自母亲,在正常人群数据库中为低频变异,根据美国医学遗传学与基因组学会发布的变异解读指南判读为疑似致病性变异,导致基因功能丧失;外显子4存在c.1619G>C错义突变,来自父亲,在正常人群数据库中为低频变异,根据美国医学遗传学与基因组学会发布的变异解读指南判读为临床意义未明,生物信息学蛋白功能预测软件SIFT、PloyPhen_2、REVEL均预测为有害。该基因为常染色体隐性遗传,经查询人类基因突变数据库,本例患儿MOGS基因突变位点为未报道过的突变。
以"先天性糖基化障碍Ⅱb型"、"先天性糖基化缺陷Ⅱb型"、"甘露糖基-低聚糖葡萄糖苷酶"为关键词检索中国知网、维普数据库和万方数据库自建库至2019年8月收录的文献,未检索到CDG-Ⅱb型相关报道。以"congenital disorders of glycosylation type Ⅱb"、"congenital defects of glycosylation type Ⅱb"、"MOGS"为关键词检索生物医学文献数据库(PubMed)、Web of Science数据库自建库至2019年8月收录的文献,共检索到相关文献4篇[4,5,6,7](6例病例),加上本文1例,共7例CDG-Ⅱb型患儿,其中男2例,女5例。7例患儿均起病早,且为多系统受累。7例中表现为面部畸形6例;神经发育迟缓、肌张力低下、喂养困难、肝大及生殖器发育不全各5例;听力异常、低通气或呼吸暂停、低丙种球蛋白血症各4例;惊厥、头颅磁共振异常各3例;叠指畸形、反复骨折、甲状腺功能减退各2例;抗利尿激素分泌异常1例;MOGS基因突变7例,均为复合杂合突变,共有11个突变位点。1岁内死亡5例,存活2例。见表1。

已报道的6例及本文1例先天性糖基化障碍Ⅱb型患儿临床资料
已报道的6例及本文1例先天性糖基化障碍Ⅱb型患儿临床资料
| 作者及发表年份 | 性别 | 起病时间 | 头面部畸形 | 神经系统 | 听力、视觉 | 呼吸系统 | 循环系统 | 消化系统 | 感染 | 内分泌 | 其他异常 | 基因突变位点 | 转归 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| De Praeter等2000[4] | 女 | 新生儿期 | 后枕部突出,眼裂小,长睫毛,阔鼻,下颌后缩,高腭弓 | 肌张力低下,惊厥,头颅磁共振正常 | 脑干反应测听及视觉诱发反应异常 | 低通气,呼吸暂停 | — | 鼻饲喂养,肝大 | 败血症 | — | 生殖器发育不全,脊柱侧弯,叠指 | c.1587G>C,c.2085T>C | 74 d死亡 |
| Sadat等2014[5] | 男 | 早期 | 面部畸形(未具体描述) | 肌张力低下,惊厥,智力障碍,脑萎缩,胼胝体小 | 感音性耳聋,视神经萎缩 | — | — | 慢性便秘 | 脓胸,骨髓炎 | — | 生殖器发育不全,反复骨折 | c.370C>T,c.65C>T,c.329G>A | 11岁存活 |
| Sadat等2014[5] | 女 | 早期 | 面部畸形(未具体描述) | 肌张力低下,惊厥,智力障碍,脑萎缩,胼胝体小 | 感音性耳聋,视神经萎缩 | — | — | 慢性便秘 | 中耳炎,尿路感染 | — | 生殖器发育不全,反复骨折 | c.370C>T,c.65C>T,c.329G>A | 6岁存活 |
| Kim等2018[6] | 男 | 新生儿期 | 多毛症,发色浅,前额狭小,眼裂小,眼距宽,长睫毛,阔鼻,下颌后缩,高腭弓 | 肌张力低下,头颅磁共振正常 | 脑干反应测听异常 | 呼吸暂停 | 房间隔缺损,左心室肥厚 | 鼻饲喂养,肝脾肿大,胆汁淤积 | — | 抗利尿激素异常分泌,甲状腺功能减退 | 生殖器发育不全,叠指 | c.2405C>T,c.1603C>T | 4月龄死亡 |
| Li等2019[7] | 女 | 4个月 | — | 运动发育迟缓;头颅磁共振示双侧额回小,胼胝体小 | 听力正常 | 呼吸浅慢 | 心功能不全 | 鼻饲喂养,肝大,肝功能不全 | — | 中枢性甲状腺功能减退 | — | c.544G>A,c.1698C>A | 9月龄死亡 |
| Li等2018[7] | 女 | 3个月 | 发色浅,前额狭小,眼裂小,鼻柱宽,高腭弓 | 运动发育迟缓 | — | — | 房间隔缺损 | 鼻饲喂养,肝大,肝功能不全 | 巨细胞病毒感染 | — | 漏斗胸,握拳 | c.544G>A,c.1698C>A | 10月龄死亡 |
| 本例患儿 | 女 | 新生儿期 | 多毛症,前囟早闭,前额狭小,长睫毛,眼裂小,眼距宽 | 肌张力低下,头颅磁共振正常 | 听力未查,眼底正常 | 低通气,呼吸暂停 | 室间隔增厚,动脉导管未闭 | 鼻饲喂养,肝大 | 肺炎 | 甲状腺功能正常 | 大阴唇发育不全 | c.1598delT,c.1619G>C | 28 d死亡 |
注:"—"为文献中未描述
CDG是由蛋白或脂肪糖基化异常所致的一组遗传代谢性疾病。当糖基化酶存在功能缺陷时,蛋白或脂肪糖基化出现异常,引起不同的临床表现[8]。现已发现100多种CDG,多与蛋白质和天门冬酰胺结合的N-连接寡聚糖合成异常有关,根据缺陷发生环节不同分为N-聚糖合成缺陷(CDG-Ⅰ型)和N-聚糖加工缺陷(CDG-Ⅱ型),根据缺陷酶的不同,其中CDG-Ⅱ型可分为a和b等多种亚型[9,10]。CDG-Ⅱb型(在线人类孟德尔遗传数据库编号606056)是由编码MOGS的基因缺陷引起的常染色体隐性遗传病。MOGS也称为葡萄糖苷酶Ⅰ,在内质网中表达,是参与N-连接寡糖加工过程的第一个酶,当MOGS严重缺乏时,阻碍后续加工步骤,最终影响N-连接聚糖的形成,引起多器官系统受累[4]。本病多起病早,常累及多系统,病情较重,进展快,寿命短,常于1岁内死亡。
不同CDG的临床表现及严重程度不同,部分症状到一定年龄才表现出来。CDG-Ⅱb型患儿临床特征相似,如异常面容(眼裂小、长睫毛、阔鼻、下颌后缩、高腭弓)、肌张力低下、惊厥、低通气、呼吸暂停、喂养困难、肝大、生殖器发育不全、低丙种球蛋白血症等。CDG-Ⅱb型常存在不同程度的神经系统受累,与蛋白质糖基化在神经细胞的黏附、迁移、突触发生和传导中的调节作用有关[11]。Sadat等[5]报道2例虽有严重低丙种球蛋白血症、但无反复感染的CDG-Ⅱb型兄妹,分析原因可能是丙种球蛋白、人类病毒受体以及病毒编码的多种蛋白质需经翻译后修饰才能发挥功能,CDG-Ⅱb型患儿因N-聚糖剪切修饰出现障碍,虽导致低丙种球蛋白血症,但同时病毒复制和进入细胞能力也减弱,降低了患儿的易感性。因此,MOGS抑制剂将来可能作为广谱抗病毒药,在抗人类免疫缺陷病毒、肝炎病毒等疾病方面存在广阔前景[12,13]。本例患儿未发生低丙种球蛋白血症,可能与入院后行该项检查较早、后续未动态检测免疫球蛋白水平有关。CDG-Ⅱb型患儿还可有甲状腺功能减退症、心功能不全、慢性便秘、反复骨折、叠指畸形等其他表型,本例患儿还存在外耳廓多毛及前囟早闭这两个新表型,由此可见,与其他CDG类型相似,CDG-Ⅱb型患儿同样存在高度异质性[7]。
转铁蛋白等电聚焦电泳(transferrin isoelectric focusing,TIEF)、尿中四糖检测、酶活性检测及基因测序等方法有助于CDG-Ⅱb型的诊断[4,5,6,7]。TIEF是CDG诊断及分型的首选检测方法。CDG-Ⅰ型、CDG-Ⅱ型患儿均有双唾液酸转铁蛋白和无唾液酸转铁蛋白含量增高,CDG-Ⅱ型患儿单唾液酸转铁蛋白和三唾液酸转铁蛋白含量也增高[6,14]。MOGS严重缺乏时,四糖产生增多,因此若检测到尿中四糖蓄积,也提示CDG-Ⅱb型[4,5]。随着基因检测的普及,通过直接应用全外显子检测确诊的CDG越来越多[14],Li等[7]报道的病例及本文病例均为直接通过基因检测确诊。
关于本病预后,Sadat等[5]报道的一对兄妹在11岁和6岁时仍存活,但均遗留智力障碍。Kane等[15]通过对2例患儿培养的成纤维细胞和外周血DNA的突变分析,发现存在野生型等位基因,并认为具有野生型等位基因的细胞其存活或生长优势改善了CDG患儿的表型,这可能是此2例患儿存活时间较长的原因。本例患儿生后28 d因家长放弃治疗死亡,住院期间肌张力低下、无法脱离呼吸机及喂养困难为主要问题,其他系统受累不明显,可能与患儿日龄小、有些表现尚未出现有关,结合文献报道病例推测其总体预后不佳。另外,国外有家族中同胞发病的报道[5,6,7]。因此,本例患儿的确诊,对指导该家长后续生育有重要意义。
尽管在过去10年里越来越多新的CDG类型被发现,但治疗进展很慢,除个别CDG亚型如CDG-Ⅰ b型和CDG-Ⅰt型等能治疗外[16,17,18,19,20],多数CDG,包括CDG-Ⅱb型目前尚无特殊治疗方法,但基因治疗、增强酶活性治疗、酶替代治疗、绕过原本代谢途径及底物补充治疗作为未来的治疗策略值得期待。
所有作者均声明不存在利益冲突

























