
Apert综合征是新生儿期罕见的遗传性疾病,因FGFR2基因变异导致,发病率1/65 000,其特征为颅缝早闭、尖头畸形、并指(趾)畸形,常合并皮肤、骨骼、大脑和其他器官异常。本文报告1例Apert综合征早产儿,以提高临床医生对此病的认识。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
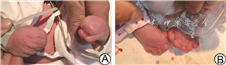
患儿男,生后6 h因“发现手足畸形”收入院。患儿系第1胎第1产,胎龄37+1周因母亲妊娠期高血压、子痫前期剖宫产娩出,出生体重2 700 g。母孕32周产检发现患儿脑室增宽,最宽1.3 mm,行无创产前基因检测提示胎儿13、18、21号染色体异常低风险,外院胎儿头颅MRI无明显异常,家长拒绝羊水穿刺,无家族遗传病史。入院查体:头颅形状略尖,有乒乓球样感,顶枕缝宽约0.7 cm,前囟2.5 cm × 2.5 cm,张力不高,双眼角略下斜,双耳耳位低,未见悬雍垂。左手腕前端可见勺状疑似手掌形态,掌前端可见3个类指状突起,无正常手指关节形态,右手腕前端可见球状突起,可触及骨性结构,无指结构;双足并趾畸形与皮肤融合,各趾末端均可见趾甲,见图1。血尿遗传代谢筛查、染色体核型分析未见异常。头颅MRI提示头颅稍尖、前后径略短,两侧脑室对称、稍宽,余未见明显异常。基因分析:FGFR2基因有1个杂合变异,758号核苷酸由胞嘧啶(C)变为鸟嘌呤(G)(c.758C>G),引起第253号氨基酸由脯氨酸变为精氨酸(p.P253R)。根据美国遗传学与基因组学学会指南,该变异初步判定为致病性变异。患儿父母基因检测该位点均无变异。诊断Apert综合征。常规监测治疗18 d,一般状况、喂养均正常,予以出院。11月龄随访,家长诉患儿不会爬,协助下可短暂独坐,神经行为发育落后于同龄儿。
Apert综合征又名尖头并指/趾畸形Ⅰ型,为常染色体显性遗传病[1],主要表现为颅缝早闭所致的头颅畸形、突眼和中面部严重发育不良,多数患儿伴有智力发育迟缓[2]。合并手和(或)足的并指(趾)畸形者,常为第2、3、4指/趾骨融合,有一个指(趾)甲,手指短小。本例患儿符合该病的临床特点。Apert综合征大部分为散发病例,其致病基因FGFR2位于染色体10q25-10q26,编码蛋白为含277个氨基酸的受体酪氨酸激酶,通过与成纤维细胞生长因子配体结合,对胚胎发育及骨骼和软骨的形成有着至关重要的作用[3]。研究显示,FGFR2基因有不同的表型,变异位点主要为S252,此位点变异患儿主要表现为严重颅面部骨畸形,P253R变异患儿的并指(趾)畸形更突出[4]。本例患儿基因变异位点为P253R,并指(趾)畸形很明显,而其父母临床无异常表现,且基因检测未见异常,考虑患儿为新发变异。Apert综合征产前超声诊断存在一定难度[5],可能与宫内胎儿足趾间毗邻紧密、双足趾较短、活动度小有关,受累程度较轻的并趾畸形超声也不易观察到。Apert综合征患儿无特异治疗方法,以治疗功能性问题为主,具体治疗方案需由多学科团队共同参与评估,并对患儿的高颅压、呼吸、视力和听力等问题进行积极随访。
所有作者均声明不存在利益冲突

























