
分析南宁市部分地区新生儿遗传代谢病疾病谱和基因谱的特征。
选择2019年7月至2021年12月在南宁市第二人民医院出生并进行多种遗传代谢病筛查的新生儿进行前瞻性研究。取新生儿足跟血后采用串联质谱法进行初筛,初筛阳性者予召回后复查串联质谱并进行生化和影像学检查,采用医学全外显子组测序技术对疑似病例进行遗传病基因检测,最后结合临床表现进行确诊,并采用Sanger测序法对确诊患儿及其父母的基因变异进行验证。
共16 207名新生儿接受遗传代谢病筛查,初筛阳性1 423例(8.8%),召回新生儿1 311例,召回率92.1%,疑似病例15例,确诊8例,总体检出率为1∶2 026。包括氨基酸代谢病4例(苯丙酮尿症2例、希特林蛋白缺乏症和酪氨酸血症各1例),有机酸代谢病2例(甲基丙二酸血症和戊二酸血症各1例),脂肪酸氧化障碍疾病2例(肉碱棕榈酰转移酶Ⅱ缺乏症和原发性肉碱缺乏各1例),其中纯合基因变异5例,分别为PAH基因2例,SLC25A13基因、SLC22A5基因、FAH基因各1例;杂合基因变异3例,分别为CPT2基因、MUT基因、GCDH基因各1例。8例患儿经规范治疗及随访,均生长发育良好。
南宁市部分地区新生儿遗传代谢病检出率较高、种类较多,及时行基因检测可早期诊断及治疗,改善患儿生存质量。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
遗传代谢病(inborn errors of metabolism,IEM)是基因变异引起酶、受体、运载蛋白或膜功能异常,从而导致机体代谢紊乱,进而产生相应病理和临床症状的一类疾病[1]。新生儿出生72 h并充分哺乳8次以后采集足跟血,采用快速、敏感的实验室方法对IEM、先天性内分泌异常等疾病进行筛查,是早期发现这类疾病的有效方法[2, 3]。本研究通过对2019年7月至2021年12月南宁市第二人民医院出生的16 207名新生儿应用血串联质谱法进行IEM筛查,分析南宁市部分地区新生儿IEM变异基因谱的特征,以期对可疑患儿能更有效、更早期的诊断及治疗,提高出生人口素质。现将结果报道如下。
选择2019年7月至2021年12月南宁市第二人民医院出生的新生儿进行前瞻性研究。入选标准:出生72 h并充分喂哺8次以上,患儿家长同意进行IEM筛查。本研究通过南宁市第二人民医院医学伦理委员会批准(KY201932),患儿家长均签署知情同意书。
1.新生儿足跟血标本采集:按照《新生儿筛查血片采集技术规范》(WS376.3-2013),采集出生72 h后并且已经充分哺乳8次以上(母乳或人工喂养均可)的新生儿足跟血(滤纸干血斑标本)。将滤纸干血斑标本水平置于干燥处晾干后送至我院检验科新生儿疾病筛查室进行串联质谱检测。
2.串联质谱筛查:(1)主要仪器和试剂:Waters TQD串联质谱分析系统(美国Waters公司)、Thermombi100-4A恒温平板振荡器(杭州奥盛公司)、涡轮震荡仪(上海沪析公司)、非衍生化多种氨基酸、肉碱和琥珀酰丙酮测定试剂盒(可筛查75种IEM);(2)检测方法:试剂孵育、提取血样标本中的氨基酸和肉碱,检测氨基酸和肉碱浓度,将氨基酸或肉碱指标异常的新生儿召回重新采集足跟血再次进行筛查,若2次筛查结果均为异常,则判断为疑似IEM患儿。
3. IEM确诊标准:对疑似新生儿IEM患儿进行召回,完善血生化及基因分析进行确诊。基因检测采用高通量Panel测序法,抽取新生儿及其父母外周血各2 ml,送至深圳联合医学科技有限公司进行IEM相关的130个基因的高通量测序。目标序列捕获采用超多重聚合酶链反应扩增的方法,覆盖所有目标基因的外显子区域及相邻的内含子区域(±50 bp)。对于可疑基因检测异常患儿采用Sanger测序法进行位点验证及父母验证,未进行拷贝数变异分析。
研究期间南宁市第二人民医院总计出生新生儿16 228名,48 h内转院或死亡21例,应用串联质谱法进行IEM筛查16 207名,初筛阳性1 423例(8.8%)。男772例(54.3%),女651例(45.7%);胎龄26~42周,早产儿378例(26.6%),足月儿1 045例(73.4%);出生体重600~4 500 g。初筛阳性患儿代谢异常分布情况见表1。
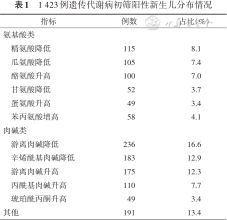
1 423例遗传代谢病初筛阳性新生儿分布情况
1 423例遗传代谢病初筛阳性新生儿分布情况
| 指标 | 例数 | 占比(%) |
|---|---|---|
| 氨基酸类 | ||
| 精氨酸降低 | 115 | 8.1 |
| 瓜氨酸降低 | 105 | 7.4 |
| 酪氨酸升高 | 100 | 7.0 |
| 甘氨酸降低 | 52 | 3.7 |
| 蛋氨酸升高 | 49 | 3.4 |
| 苯丙氨酸增高 | 58 | 4.1 |
| 肉碱类 | ||
| 游离肉碱降低 | 236 | 16.6 |
| 辛烯酰基肉碱降低 | 183 | 12.9 |
| 游离肉碱升高 | 175 | 12.3 |
| 丙酰基肉碱升高 | 110 | 7.7 |
| 琥珀酰丙酮升高 | 49 | 3.4 |
| 其他 | 191 | 13.4 |
召回复筛新生儿1 311例,召回率92.1%,复筛阳性33例,其中86.6%使用左卡尼汀的早产儿停药后复查异常指标已降为正常,对复筛阳性患儿再进一步根据生化检验、神经系统检测及基因检测,确诊IEM患儿8例,即本研究地区、本研究时段内新生儿IEM检出率为1∶2 026。确诊病例情况见表2。
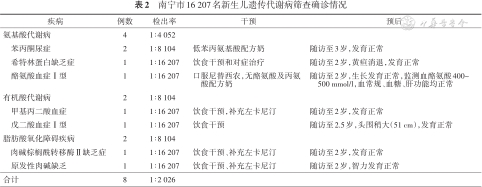
南宁市16 207名新生儿遗传代谢病筛查确诊情况
南宁市16 207名新生儿遗传代谢病筛查确诊情况
| 疾病 | 例数 | 检出率 | 干预 | 预后 |
|---|---|---|---|---|
| 氨基酸代谢病 | 4 | 1∶4 052 | ||
| 苯丙酮尿症 | 2 | 1∶8 104 | 低苯丙氨基酸配方奶 | 随访至3岁,发育正常 |
| 希特林蛋白缺乏症 | 1 | 1∶16 207 | 饮食干预和对症治疗 | 随访至2岁,黄疸消退,发育正常 |
| 酪氨酸血症Ⅰ型 | 1 | 1∶16 207 | 口服尼替西农,无酪氨酸及丙氨酸配方奶 | 随访至2岁,生长发育正常,监测血酪氨酸400~500 mmol/l,血常规、血糖、肝功能均正常 |
| 有机酸代谢病 | 2 | 1∶8 104 | ||
| 甲基丙二酸血症 | 1 | 1∶16 207 | 饮食干预,补充左卡尼汀 | 随访至2岁,发育正常 |
| 戊二酸血症Ⅰ型 | 1 | 1∶16 207 | 饮食干预 | 随访至2.5岁,头围稍大(51 cm),发育正常 |
| 脂肪酸氧化障碍疾病 | 2 | 1∶8 104 | ||
| 肉碱棕榈酰转移酶Ⅱ缺乏症 | 1 | 1∶16 207 | 饮食干预、补充左卡尼汀 | 随访至2岁,发育正常 |
| 原发性肉碱缺乏 | 1 | 1∶16 207 | 饮食干预、补充左卡尼汀 | 随访至2岁,智力发育正常 |
| 合计 | 8 | 1∶2 026 |
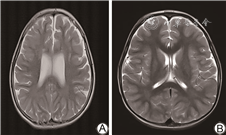
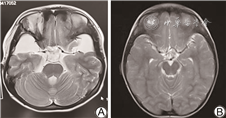
电话回访112例未同意复筛的患儿,其中拒接电话22例,接受电话回访的新生儿家长均回复婴儿生后至今(随访年龄0~3岁)健康。对8例确诊IEM患儿进行追踪随访及智力评估(随访年龄0~3岁),1例甲基丙二酸血症患儿早期头颅MRI显示双侧苍白球区、双侧尾状核头对称片状异常信号;1例戊二酸血症患儿早期头颅MRI提示额叶白质内等T1长T2信号,提示双侧额叶白质脱髓鞘。所有确诊病例均给予对应的治疗,包括特殊奶粉、补充左卡尼汀、维生素B12及尼替西农等,目前均生长发育良好。其中甲基丙二酸血症患儿随访至2岁、戊二酸血症患儿随访至2.5岁时头颅MRI均提示正常,智力正常。见图1、2。
疑似IEM患儿中有33例进行了基因检测,其中8例检出相关基因变异,肉碱棕榈酰转移酶Ⅱ缺乏症、甲基丙二酸血症、戊二酸血症Ⅰ型各1例,上述位点均经父母验证确认为复合杂合变异。其他5例为纯合变异,均符合常染色体隐性遗传病的遗传特征。见表3。
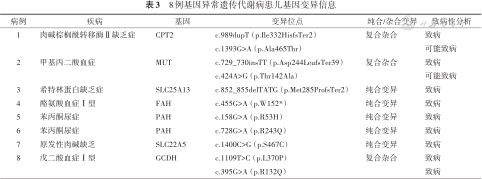
8例基因异常遗传代谢病患儿基因变异信息
8例基因异常遗传代谢病患儿基因变异信息
| 病例 | 疾病 | 基因 | 变异位点 | 纯合/杂合变异 | 致病性分析 |
|---|---|---|---|---|---|
| 1 | 肉碱棕榈酰转移酶Ⅱ缺乏症 | CPT2 | c.989dupT(p.Ile332HisfsTer2) | 复合杂合 | 致病 |
| c.1393G>A(p.Ala465Thr) | 可能致病 | ||||
| 2 | 甲基丙二酸血症 | MUT | c.729_730insTT(p.Asp244LeufsTer39) | 复合杂合 | 致病 |
| c.424A>G(p.Thr142Ala) | 可能致病 | ||||
| 3 | 希特林蛋白缺乏症 | SLC25A13 | c.852_855delTATG(p.Met285ProfsTer2) | 纯合变异 | 致病 |
| 4 | 酪氨酸血症Ⅰ型 | FAH | c.455G>A(p.W152*) | 纯合变异 | 致病 |
| 5 | 苯丙酮尿症 | PAH | c.158G>A(p.R53H) | 纯合变异 | 致病 |
| 6 | 苯丙酮尿症 | PAH | c.728G>A(p.R243Q) | 纯合变异 | 致病 |
| 7 | 原发性肉碱缺乏 | SLC22A5 | c.1400C>G(p.S467C) | 纯合变异 | 致病 |
| 8 | 戊二酸血症Ⅰ型 | GCDH | c.1109T>C(p.L370P) | 复合杂合 | 致病 |
| c.395G>A(p.R132Q) | 致病 |
IEM是由于基因变异导致细胞膜功能异常、酶缺陷或受体缺陷造成的各种代谢障碍,如不及时干预可能危及生命[1]。国内报道显示,新生儿IEM总体初筛阳性率为0.6%~1.2%,总体发病率为1∶8 304~1∶1 545[2, 3, 4]。本研究应用串联质谱技术对广西南宁市第二人民医院出生的新生儿进行IEM筛查,初筛阳性率为8.8%,明显高于国内其他研究,总体检出率为1∶2 026,处于较高水平。
本研究确诊氨基酸代谢病4例,占确诊病例数的50.0%,包括苯丙酮尿症2例,希特林蛋白缺乏症和酪氨酸血症Ⅰ型各1例。2例苯丙酮尿症患儿均为纯合基因变异,变异基因为c.728G>A、c.158G>A,符合经典变异[5],经早期服用低苯丙氨酸奶粉,现随访至3岁,智力及运动均发育良好,因此,早发现、早干预可避免患儿神经系统损伤[6]。1例希特林蛋白缺乏症患儿以持续性黄疸为临床表现,SLC25A13基因纯合移码变异c.852_855delTATG(p.Met285ProfsTer2),为致病性变异[7, 8],予无乳糖和富含中链脂肪酸奶粉,患儿目前已2岁,生长发育良好。1例患儿因筛查发现酪氨酸、琥珀酰丙酮明显增高,基因检测为FAH基因c.455G>A纯合变异,诊断酪氨酸血症Ⅰ型,予不含苯丙氨酸、酪氨酸的特殊奶粉结合母乳混合喂养,并予尼替西农口服治疗[9],定期随访评估体格及智力等生长发育情况,监测血清酪氨酸水平及血常规、血糖、肝功能,已随访至2岁,智力及发育正常。
本研究确诊甲基丙二酸血症1例,串联质谱筛查提示甲基丙二酸及丙酰肉碱明显增高,基因检测MUT基因c.729_730insTT(p.Asp244LeufsTer39)及c.424A>G(p.Thr142Ala)复合杂合变异,可能影响底物结合区和钴胺素结合区功能[10, 11],为致病变异。头颅MRI提示双侧额叶白质脱髓鞘,侧脑室轻度扩张,经维生素B12、左卡尼汀治疗,并予以饮食干预,临床症状消失,随访至2岁,韦氏量表测试智力正常,复查MRI正常。本研究确诊戊二酸血症Ⅰ型1例,该病可能导致大头畸形、肌张力异常及运动障碍等神经系统发育异常[12],该患儿血串联质谱提示戊二酰肉碱、戊二酰肉碱/辛酰肉碱水平升高,头颅MRI提示双侧颞部蛛网膜下腔明显扩大,基因检测及家系验证为GCDH基因c.1109T>C(p.L370P)及c.395G>A(p.R132Q)复合杂合变异,变异位点分别来源于父母,予口服维生素B2及左卡尼汀,随访到2岁半,复查头颅MRI正常,头围稍大,无神经系统发育障碍。
本研究确诊脂肪酸氧化障碍疾病2例,其中1例为肉碱棕榈酰转移酶Ⅱ缺乏症,该病临床可表现为低血糖、昏迷、癫痫、肌病以及肝功能异常等[13, 14],本例患儿CPT2基因c.989dupT(p.Ile332HisfsTer2)及c.1393G>A(p.Ala465Thr)复合杂合变异,既往未见研究报道,为新发变异位点。该患儿通过补充左卡尼汀维持血中游离肉碱水平稳定等治疗,随访发育正常。另1例为原发性肉碱缺乏,该患儿原发性肉碱C0降至2.12 μmol/L,基因检测结果提示SLC22A5基因纯合变异c.1400C>G(p.S467C),为变异热点[15],经口服左卡尼汀,现随访至2岁,游离肉碱维持正常,生长发育及智力正常。
本次新生儿疾病筛查的初筛阳性率较高,召回工作量大,这是进行新生儿IEM筛查的难题[16]。初筛阳性率明显高于国内其他研究的原因有:(1)本研究中游离肉碱增高175例,占12.3%,占比较高,考虑为早产儿早期使用左卡尼汀有关,停药后复查绝大数降为正常。(2)部分新生儿吃奶欠佳,家长未在规定时间内充分哺乳,没有摄入足够蛋白质,血中代谢产物低而出现筛查结果异常[17]。(3)部分筛查阳性者为重症患儿,病情好转后复筛降至正常,考虑初筛阳性与患儿体内代谢紊乱有关[18]。
多数IEM新生儿早期临床表现不明显,对初筛阳性患儿需及时召回复筛,通过串联质谱筛查联合基因检测确诊南宁市第二人民医院IEM共8例,经早期有效的治疗,多数预后良好。对确诊病例开展父母及患儿的IEM基因检测,对指导家庭成员遗传咨询及今后的产前诊断有重要指导意义,及时进行串联质谱筛查新生儿IEM有利于控制出生缺陷的发生和提高出生人口质量。
覃晓, 旷娟, 蓝国锋, 等. 南宁市部分地区新生儿遗传代谢病疾病谱和基因谱特征分析[J]. 中华新生儿科杂志, 2023, 38(5): 289-293. DOI: 10.3760/cma.j.issn.2096-2932.2023.05.007.
所有作者声明无利益冲突

























