
本文报道1例新生儿Cantú综合征,患儿生后因水肿、青紫入院,伴多毛、特殊面容,全外显子组测序发现患儿ABCC9基因c.3605C>T位点杂合变异,随访患儿存在智力发育落后。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿女,因“生后水肿、呻吟44 min”入院。患儿系第3胎第2产,胎龄37+5 周因“母亲瘢痕子宫”剖宫产娩出,出生体重3 600 g,出生史无异常。母孕期唐氏筛查高风险,NT增厚,无创DNAplus低风险,孕37+2周B超示羊水指数19.6 cm,羊水偏多。母亲血型A型,Rh(D)阳性,妊娠期无糖尿病、高血压等并发症。入院查体:身长57 cm(>P90),头围38 cm(>P90),体重3 600 g(>P90)。口周青紫,吐沫、呻吟,鼻梁低、舌大,前额、颞部毛发浓密,腰臀部体毛增多,全身水肿,头皮、颜面、腹壁、会阴部尤甚,双肺可闻及粗湿啰音,心腹查体无异常,四肢末梢稍凉,肌张力低,原始反射减弱。血气分析:pH 7.33,PCO2 32.2 mmHg,PO2 67.6 mmHg,BE-9.31 mmol/L。血常规、超敏C反应蛋白、降钙素原、甲状腺功能、肝肾功能、心肌酶谱均正常,G6PD酶活性正常,TORCH IgM阴性,Coombs试验阴性,血培养阴性。胸部X线片示双肺少量渗出病灶。心脏超声示动脉导管未闭(patent ductus arteriosus,PDA),房间隔缺损(1.5 mm),卵圆孔未闭(3.3 mm),左房稍大,肺动脉收缩压40 mmHg。头颅MRI、腹部及胃肠道超声未见异常。NBNA评分35分。染色体核型分析46,XX。入院后予持续气道正压辅助通气、头孢噻肟钠联合青霉素抗感染等治疗,呼吸好转,尿量正常,全身水肿无明显消退,生后3 d行家系全外显子基因检测;住院期间患儿吸吮欠佳,自行进乳慢,会阴部持续水肿,生后9 d家属要求出院。生后37 d基因检测回报:患儿ABCC9外显子区域c.3605C>T杂合变异,患儿父母均未携带该变异,为新发变异(图1),HGMD数据库、ACMG指南归类为致病,Cinvar数据库收录为可能致病,诊断Cantú综合征(Cantú syndrome,CS)。院外定期复查心脏超声,示动脉导管已关闭,房间隔缺损(多发孔型),右心房、右心室扩大;1岁左右行“房间隔缺损+三尖瓣修补术”,手术顺利。现1岁余,水肿消退,仍有多毛、发育迟缓,0~6岁智力发育筛查量表测试DQ<70,复查心脏超声示房间隔缺损修补术后,二、三尖瓣轻度关闭不全;肺动脉收缩压55 mmHg,少量心包积液。现口服波生坦、呋塞米、螺内酯治疗中。
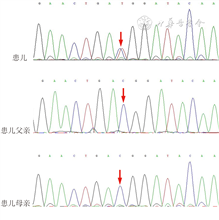
CS是一种罕见的常染色体显性遗传病,由编码ATP敏感钾通道(K-ATP)亚单位的KCNJ8或ABCC9基因变异引起,其中ABCC9基因变异更常见;临床特征为先天性多毛症、大头及面部畸形、骨骼及多种心脑血管异常[1]。CS在1982年由Cantú首次报道[1],总体发病率未见报道,目前国内报道过3例[2],国外最大的CS队列研究纳入了74例[3]。CS患者半数以上在胎儿期出现羊水过多,新生儿期表现为巨大儿、大头畸形、特殊面容、多毛、水肿、皮肤褶皱或松弛、心脏畸形、脑血管异常、骨软骨发育不良[1,3]。多数CS患者婴幼儿期存在肌力减退、生长发育迟缓、发育里程碑延迟,部分早期需康复治疗,部分青春期可出现多动症、自闭症等行为问题,但持续的智力障碍并不明显,多数可达到正常教育水平[4]。本例患儿胎儿期羊水多,生后有特殊面容、多毛、大头畸形、全身水肿、呼吸困难,全外显子组测序发现ABCC9基因变异,最终诊断CS。
约80%的CS患者存在心脏异常,部分可发展为心功能不全伴肺动脉高压、二尖瓣反流/功能不全、三尖瓣反流/功能不全[3]。目前尚无针对CS的靶向治疗方法,也不清楚心血管异常是否可被逆转。研究证实K-ATP通道激活是CS患者发病的基础[5],因此K-ATP通道抑制剂(如格列本脲)可能用于CS的治疗。Ma等[6]报道过1例使用格列本脲治疗的CS早产儿,患儿生后存在PDA、持续性肺动脉高压,常规治疗后呼吸状况改善不明显,生后11周开始格列本脲试验剂量治疗[0.05 mg/(kg·d),分2次],1周后患儿呼吸、水肿改善,仅在夜间睡眠时使用无创通气,13月龄时格列本脲加量至0.15 mg/(kg·d),治疗期间患儿曾出现短暂性轻度低血糖。由于格列本脲治疗CS经验有限、安全剂量不明及低血糖等不良反应,有待进一步研究。本例患儿生后早期存在PDA、房间隔缺损、卵圆孔未闭,常规治疗后呼吸症状缓解,后行心脏手术治疗,现存在多毛、发育迟缓,还需长期随访。
当遇到特殊面容、多毛、心脏畸形、持续水肿的患儿,应尽早行基因检测明确诊断。因CS患儿多毛、水肿症状持久存在,心脑血管畸形可进展为心衰、脑卒中,部分存在生长发育落后,需与家属做好沟通,早期干预以改善预后,并做好长期随访。
何明嫄, 聂潘荣. 新生儿Cantú综合征1例[J]. 中华新生儿科杂志, 2023, 38(5): 304-306. DOI: 10.3760/cma.j.issn.2096-2932.2023.05.011.
所有作者声明无利益冲突

























